UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
(Mark One)
|
|
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended:
March 31, 2007
|
|
OR
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to .
|
Commission File Number:
001-33216
OCULUS INNOVATIVE SCIENCES,
INC.
(Exact name of Registrant as
specified in its charter)
| |
|
|
|
Delaware
|
|
68-0423298
|
(State or other jurisdiction
of
incorporation or organization)
|
|
(I.R.S. Employer
Identification Number)
|
|
|
|
|
1129 N. McDowell
Blvd.
Petaluma, California
(address of principal
executive offices)
|
|
94954
(zip
code)
|
(707) 782-0792
(Registrant’s telephone
number, including area code)
None
(Securities registered pursuant
to Section 12(b) of the Act)
Common Stock
(Securities registered pursuant
to Section 12(g) of the Act and Title of Class)
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 15(d) of the Act.
Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. þ
Indicate by check mark whether registrant is a large accelerated
filer, an accelerated filer, or a non-accelerated filer. See
definition of “accelerated filer and large accelerated
filer” in
Rule 12b-2
of the Exchange Act. (check one):
Large accelerated
filer o Accelerated
filer o Non-accelerated
filer þ
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
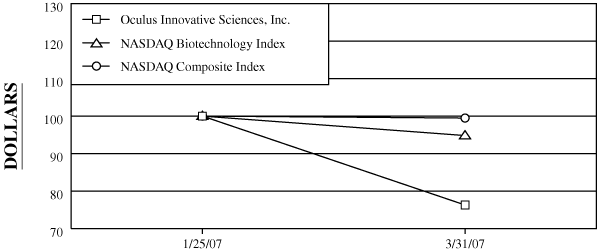
The registrant’s common stock first traded on the Nasdaq
Global Market on January 25, 2007. Accordingly, the
registrant’s common stock was not trading publicly on
September 30, 2006. As of May 31, 2007, the aggregate
market value of voting and non-voting common stock held by
non-affiliates of the registrant was approximately
$74.2 million, based on the closing price of the common
stock as reported on the Nasdaq Global Market for that date.
There were 11,844,411 shares of the registrant’s
Common Stock issued and outstanding on May 31, 2007.
DOCUMENTS
INCORPORATED BY REFERENCE
Items 10, 11, 12, 13 and 14 of Part III incorporate by
reference information from the registrant’s proxy statement
to be filed with the Securities and Exchange Commission in
connection with the solicitation of proxies for the
registrant’s 2007 Annual Meeting of Stockholders to be held
on September 30, 2007.
PART I
This Report contains forward-looking statements within the
meaning of the Private Securities Litigation Reform Act of 1995.
When used in this Report, the words “expects,”
“anticipates,” “intends,”
“estimates,” “plans,” “projects,”
“continue,” “ongoing,”
“potential,” “expect,” “predict,”
“believe,” “intend,” “may,”
“will,” “should,” “could,”
“would” and similar expressions are intended to
identify forward-looking statements. These are statements that
relate to future periods and include statements about, but not
limited to: the progress and timing of our development programs
and regulatory approvals for our products; the benefits and
effectiveness of our products; the development of protocols for
clinical studies; enrollment in clinical studies; the progress
and timing of clinical trials and physician studies; our
expectations related to the use of our cash reserves; our
ability to manufacture sufficient amounts of our product
candidates for clinical trials and products for
commercialization activities; the outcome of discussions with
the FDA and other regulatory agencies; the content and timing of
submissions to, and decisions made by, the FDA and other
regulatory agencies, including demonstrating to the satisfaction
of the FDA the safety and efficacy of our products; the ability
of our products to meet existing or future regulatory standards;
the rate and causes of infection; the accuracy of our estimates
of the size and characteristics of the markets which may be
addressed by our products; our expectations and capabilities
relating to the sales and marketing of our current products and
our product candidates; our ability to penetrate markets through
our sales force, distribution network, and strategic
business partners and generate attractive margins; the expansion
of our sales force and distribution network; the establishment
of strategic partnerships for the development or sale of
products; the ability to attain specified revenue goals within a
specified time frame, if at all, or to reduce costs; the timing
of commercializing our products; our ability to protect our
intellectual property and operate our business without
infringing on the intellectual property of others; our ability
to continue to expand our intellectual property portfolio; our
expectations about the outcome of litigation and controversies
with third parties; our ability to attract and retain qualified
directors, officers and employees; our relationship with Quimica
Pasteur; our ability to compete with other companies that are
developing or selling products that are competitive with our
products; the ability of our products to become the standard of
care for controlling infection in chronic and acute wounds; our
ability to expand to and commercialize products in markets
outside the wound care market; our estimates regarding future
operating performance, earnings and capital requirements; our
expectations with respect to our microbiology contract testing
laboratory; our expectations relating to the concentration of
our revenue from international sales; and the impact of the
Sarbanes-Oxley Act of 2002 and any future changes in accounting
regulations or practices in general with respect to public
companies.
Forward-looking statements are subject to risks and
uncertainties that could cause actual results to differ
materially from those projected. These risks and uncertainties
include, but are not limited to, those risks discussed below, as
well as our ability to develop and commercialize new products;
the risk of unanticipated delays in research and development
efforts; the risk that we may not obtain reimbursement for our
existing test and any future products we may develop; the risks
and uncertainties associated with the regulation of our products
by the U.S. Food and Drug Administration; the ability to
compete against third parties; our ability to obtain capital
when needed; our history of operating losses and the risks set
forth under “Risks Related to our Business.” These
forward-looking statements speak only as of the date hereof. The
Company expressly disclaims any obligation or undertaking to
release publicly any updates or revisions to any forward-looking
statements contained herein to reflect any change in the
Company’s expectations with regard thereto or any change in
events, conditions or circumstances on which any such statement
is based.
Corporate
Information
We were incorporated in California in 1999 as Micromed
Laboratories, Inc. In August 2001, we changed our name to Oculus
Innovative Sciences, Inc. In December 2006, we reincorporated in
Delaware. Our principal executive offices are located at
1129 N. McDowell Blvd., Petaluma, California, 94954,
and our telephone number is
(707) 782-0792.
We have two principal subsidiaries: Oculus Technologies of
Mexico, S.A. de C.V., organized in Mexico, and Oculus Innovative
Sciences Netherlands, B.V., organized in The Netherlands. We
also have a subsidiary, Oculus Innovative Sciences Japan, KK.,
organized under Japanese law. Our website is
www.oculusis.com.
1
Overview
We have developed, and we manufacture and market, a family of
products intended to prevent and treat infections in chronic and
acute wounds. Infection is a serious potential complication in
both chronic and acute wounds, and controlling infection is a
critical step in wound healing. Our platform technology, called
Microcyn, is a proprietary oxychlorine small molecule
formulation that is designed to treat a wide range of organisms
that cause disease, or pathogens, including viruses, fungi,
spores and antibiotic resistant strains of bacteria, such as
Methicillin-resistant Staphylococcus aureus, or MRSA, and
Vancomycin-resistant Enterococcus, or VRE, in wounds. We
do not have the necessary regulatory approvals to market
Microcyn in the United States as a drug, nor do we have the
necessary regulatory clearance or approval to market Microcyn in
the U.S. as a medical device for an antimicrobial or wound
healing indication. However, our device product is cleared for
sale in the United States as a medical device for wound
cleaning, or debridement, lubricating, moistening and dressing;
is a device under CE Mark in Europe; and is approved as a drug
in India and Mexico.
Clinical testing we conducted in connection with our submissions
to the FDA, as well as physician clinical studies, suggest that
our Microcyn-based product may help reduce a wide range of
pathogens from acute and chronic wounds. These physician
clinical studies suggest that our Microcyn-based product is easy
to use and complementary to most existing treatment methods in
wound care. Physician clinical studies in the United States
suggest that our 510(k) product may shorten hospital stays,
lower aggregate patient care costs and, in certain cases, reduce
the need for system-wide, or systemic, antibiotics.
In 2005, chronic and acute wound care represented an aggregate
of $9.6 billion in global product sales, of which
$3.3 billion was spent for the treatment of skin ulcers,
$1.6 billion to treat burns and $4.7 billion for the
treatment of surgical and trauma wounds, according to Kalorama
Information, a life sciences market research firm. We believe
our addressable market for the treatment of skin ulcers is
approximately $1.3 billion, $300 million for the
treatment of burns and $700 million for the treatment of
surgical and trauma wounds. Common methods of controlling
infection, including topical antiseptics and antibiotics, have
proven to be only moderately effective in combating infection in
the wound bed. However, topical antiseptics tend to inhibit the
healing process due to their toxicity and may require
specialized preparation or handling. Antibiotics can lead to the
emergence of resistant bacteria, such as MRSA and VRE. Systemic
antibiotics may not be effective in controlling infection in
patients with disorders affecting circulation, such as diabetes,
which are commonly associated with chronic wounds. As a result,
no single treatment is used across all types of wounds and
stages of healing.
We believe Microcyn provides significant advantages over current
methods of care in the treatment of a wide range of chronic and
acute wounds throughout all stages of treatment. These stages
include cleaning, or debridement, prevention and treatment of
infections and wound healing. We believe that Microcyn may be
the first topical product that is effective against a broad
range of bacteria and other infectious microbes including
antibiotic resistant strains such as MRSA and VRE, without
causing irritation of or damage to healthy tissue. Unlike most
antibiotics, we believe Microcyn does not target specific
strains of bacteria, a practice which has been shown to promote
the development of resistant bacteria. In addition, our products
are shelf stable, require no special preparation, and are easy
to use.
Our goal is to become a worldwide leader in anti-infectives in
treating wounds. We currently have, and intend to seek
additional regulatory clearances and approvals to market our
Microcyn-based products worldwide. In July 2004, we began
selling Microcyn in Mexico after receiving approval from the
Mexican Ministry of Health, or MOH, for the use of Microcyn as
an antiseptic, disinfectant and sterilant. Since then,
physicians in the United States, Europe, India and Mexico have
conducted 21 physician clinical studies assessing
Microcyn’s use in the treatment of infections in a variety
of wound types, including hard-to-treat wounds such as diabetic
ulcers and burns. These studies were not intended to be
rigorously designed or controlled clinical trials and, as such,
did not have all of the controls required for clinical trials
used to support an NDA submission to the FDA in that they did
not include blinding, randomization, predefined clinical end
points, use of placebo and active control groups or
U.S. good clinical practices requirements. We used the data
generated from some of these studies to support our application
for the CE Mark, or European Union certification, for wound
cleaning and reduction of infection. We received the CE Mark in
November 2004 and additional international approvals in Canada,
Mexico and India. Microcyn has also received three FDA 510(k)
2
clearances for use as a medical device in wound cleaning, or
debridement, lubricating, moistening and dressing, including
traumatic wounds and acute and chronic dermal lesions.
In the second quarter of 2007, we initiated a Phase II
randomized clinical trial, which is designed to evaluate the
effectiveness of Microcyn in mildly infected diabetic foot
ulcers with endpoints of resolution of all symptoms of
inflammation, or clinical cure, and improvement in signs
and symptoms of infection supported by microbiological response
as described in FDA guidelines. We are using more than 10
clinical sites with a target of enrolling 60 patients in
three arms using Microcyn alone, Microcyn plus an oral
antibiotic and saline plus an oral antibiotic. We expect to
announce the results of our Phase II trial in autumn of
2007. A well known contract research organization is
coordinating, monitoring and documenting results of this trial.
Following the completion of this trial, and a review meeting
with the FDA, we intend to initiate two Phase III trials.
We anticipate that patient enrollment for Phase III trials
will start in early 2008, and the trials will last about 12 to
18 months. These Phase II and Phase III clinical
trials are intended to provide the clinical basis for submission
to the FDA of a new drug application, or NDA, for the treatment
of infected diabetic foot ulcers. In the event that we obtain
drug approval from the FDA, we may seek clearance for treatment
of other types of wounds. We are currently pursuing strategic
partnerships to assess potential applications for Microcyn in
several other markets, including respiratory, ophthalmology,
dermatology, dental and veterinary markets, and FDA or other
governmental approvals may be required for any potential new
products or new indications. We have reduced expenses in our
international operations in order to focus our resources on our
U.S. clinical trials.
We currently make Microcyn available under our 510(k) clearances
in the United States primarily through our website, one national
distributor and several regional distributors. We plan for a
more aggressive commercialization and product launch in the
event we obtain drug approval from the FDA. Most of our current
marketing efforts in the United States are designed to build
brand awareness. In Europe, we sell Microcyn through exclusive
distribution agreements with distributors, all of which, we
believe, are experienced suppliers to hospitals, supported by a
distributor coordinator. We are seeking a significant
distribution partner to sell the product in Europe into the
wound care market. Also, we have a distribution agreement with a
private company in Europe, that distributes Microcyn in Europe
to salons for cleaning hands and feet during treatment. In
Mexico, we sell Microcyn through a network of distributors and
through a contract sales force, including salespeople, nurses
and clinical support staff. In India we sell through Alkem, the
6th largest pharmaceutical company in India. This year is
the first full year of the product launch of Microcyn in India.
In China, we recently signed a distribution agreement with China
Bao Tai, which intends to distribute Microcyn to hospitals,
doctors and clinics through Sinopharm, the largest
pharmaceutical company in China, and to retail pharmacies
through Lianhua Supermarkets after required regulatory approval
in China is obtained.
Our goal is to achieve the following milestones through calendar
2009:
2007
|
|
|
| |
•
|
Initiate and complete Phase II trial for use of Microcyn in
the treatment of infections in mildly infected diabetic foot
ulcers
|
| |
| |
•
|
Complete a meeting with FDA regarding our Phase II results
and Phase III protocols
|
| |
| |
•
|
Initiate site selection and investigation review board, or IRB,
approvals for Phase III clinical trial for Microcyn in the
treatment of infections in mildly infected diabetic foot ulcers
|
| |
| |
•
|
Execute distribution/partnership agreements for Microcyn outside
of the United States
|
| |
| |
•
|
Initiate partner discussions on Microcyn involving non-wound
care applications
|
| |
| |
•
|
File additional patents on new formulations and drug delivery
systems
|
2008
|
|
|
| |
•
|
Initiate Phase III trials for evaluating Dermacyn’s
effectiveness for treatment of infection in infected diabetic
foot ulcers
|
3
|
|
|
| |
•
|
Execute additional partnerships for commercialization of
Microcyn in non-wound care applications
|
| |
| |
•
|
File and obtain additional patents on new formulations and drug
delivery systems
|
2009
|
|
|
| |
•
|
Data expected from Phase III clinical trials for Microcyn
in the treatment of infections in foot ulcers
|
| |
| |
•
|
File an NDA with the FDA for treatment of infected foot ulcers
|
We cannot guarantee that we will obtain on a timely basis, if at
all, the necessary FDA approval to market Microcyn in the United
States for the treatment of infection in diabetic foot ulcers. A
number of factors can delay or prevent completion of human
clinical trials, particularly patient recruitment. Moreover,
many drug candidates fail to successfully complete clinical
trials. After an NDA is filed with the FDA, the FDA commences an
in-depth review of the NDA that takes ten months to a year to
complete but may take longer. In addition, we cannot guarantee
that we will obtain on a timely basis, or at all, the necessary
510(k) clearances for the next generation Microcyn product
formulation. The milestones described above assume that we have
sufficient funds to complete, and that we do complete, our
clinical trials for the treatment of infection in mildly
infected diabetic foot ulcers, that we have sufficient funds to
conduct Phase III trials, that the results from these
clinical trials support an NDA filing and that our products will
be commercially viable. We cannot guarantee that we will find
appropriate distribution or strategic partners, generate revenue
sufficient to fund our cash flow needs or that we will meet any
of the milestones described above in a timely manner or at all.
We also operate a microbiology contract testing laboratory
division that provides consulting and laboratory services to
medical companies that design and manufacture biomedical devices
and drugs, as well as testing on our products and potential
products. Our testing laboratory complies with U.S. good
manufacturing practices and quality systems regulation. We are
in the process of transitioning our business away from providing
laboratory services to others, as we continue to focus our
efforts on completion of our clinical trials.
Industry
Background
Wound
Care Industry Overview
According to Medtech Insight, a Division of Windhover
Information, there were over 90 million incidents of wounds
in the United States during 2004. Of these, over six million
were chronic wounds, including arterial, diabetic, pressure and
venous ulcers. The remaining 84 million were acute wounds,
which follow the normal process of healing and commonly include
burns, traumatic wounds, and approximately 67 million
surgical incisions.
Key trends in wound care include:
|
|
|
| |
•
|
large and increasing elderly, diabetic and obese populations,
each of which is vulnerable to developing a variety of
difficult-to-heal ulcers;
|
| |
| |
•
|
increased emphasis on controlling the cost of patient care in
hospitals, wound care centers and in private practice;
|
| |
| |
•
|
technological innovation, which has expanded treatment options
from traditional ointments and gauze to include advanced
treatments, such as vacuum devices, silver dressings, ultrasound
and skin grafts;
|
| |
| |
•
|
increased focus on improving the patient experience, including
reduction of pain and accelerated healing time; and
|
| |
| |
•
|
adjunctive nature of the market where multiple treatment methods
are employed, either simultaneously or sequentially, depending
on the type and stage of the wound.
|
Wound care is complex, and controlling infection is a critical
step in wound healing. Difficult-to-heal wounds can result from
traumatic injury, diabetes, peripheral vascular disease,
complications following surgery, rheumatoid arthritis,
congestive heart failure, arterial or venous ulcers and many
other conditions which compromise
4
circulation. Without proper medical intervention and control of
infection, these types of wounds typically remain open and
chronically infected.
Chronic Wounds
Chronic wounds are wounds that do not heal within a normally
expected time frame under standard care. The most frequently
occurring chronic wounds are venous, arterial, pressure and
diabetic foot ulcers. According to Medtech Insight, in 2004, the
incidence of chronic wounds in the United States was
approximately 6.1 million, comprised of 2.0 million
pressure ulcers, 1.7 million arterial ulcers,
1.6 million venous ulcers and 800,000 diabetic foot ulcers.
In addition to being expensive to treat, chronic wounds are
debilitating, painful and can result in amputations and other
serious consequences. Clinical studies suggest that, depending
on the severity of the wound, up to 43% of patients with
diabetic foot ulcers undergo an amputation. Furthermore, the
five year survival rate for patients undergoing amputations as a
result of diabetic foot ulcers is 27%.
The increasing prevalence of chronic wounds is driven by the
large and growing elderly, diabetic and obese populations.
Aging. People aged 65 and over are more
susceptible to wounds that become chronic than the overall
population. In 2006, there were more than 37 million people
in the United States over 65, representing more than 12% of the
population. By 2030, this group is expected to comprise more
than 19% of the total population of the United States, according
to U.S. Census Bureau projections. Additionally, according
to Medtech Insight, 70% of pressure ulcers occur in people
age 70 years or older, and 25% of patients in nursing
homes suffer from pressure ulcers.
Diabetes. Diabetics are particularly
vulnerable to chronic wounds as a result of the debilitating
effect of diabetes on the circulatory system. According to the
Centers for Disease Control and Prevention, or CDC, one out of
three children born in 2000 in the United States will develop
diabetes. In 2004 there were approximately 14.7 million
diabetic Americans, representing 5% of the total population, up
from 2.7% in 1990. Furthermore, according to the CDC, the
incidence of diabetes is significantly higher in people over 65:
in 2004, 16% of people over 65 were diabetic compared to 7.5% of
the total population.
Obesity. Obesity is a leading cause of Type
II, or “adult onset,” diabetes, making the obese
population more likely to eventually sustain chronic wounds.
Obesity in the United States is a growing problem. According to
the National Institute of Diabetes and Digestive and Kidney
Diseases, more than 30% of the United States adult population
was obese in 2000, up from 13% in 1960.
Acute
Wounds
Acute wounds are typically caused by traumatic injury or
surgical incision and are broadly categorized as those that can
be expected to heal within a definable timeframe. However, the
healing process may be affected by complicating factors such as
infection, leading to chronic wounds.
All acute wounds have the potential for infection and may
require prophylactic treatment to prevent infection. According
to Medtech Insight, in 2004, about 16.2 million traumatic
wounds were treated, including 8.7 million open wounds.
Also according to Medtech Insight, in 2004, approximately
67 million surgical wounds were reported in the United
States, including 36 million completed under anesthesia.
Despite modern infection control procedures, and technologies at
hospitals and surgery centers, every time the skin is opened
there is a risk of infection. We believe that there is a higher
likelihood of infection in surgeries involving anesthesia
because of the length of time the wound is open. In a clinical
study on surgical infections, it was shown that infection rates
vary with the time required to complete the surgery. For
example, infection rates varied from about 3.6% for surgeries
taking less than 30 minutes to about 16.4% for those longer than
5 hours.
Critical
Steps for Wound Treatment
Infection
Control
According to the Committee to Reduce Infection Deaths, or RID,
one out of every 20 patients contracts an infection while
in the hospital. Certain infections are increasingly dangerous
because they cannot be effectively
5
controlled by commonly used antibiotics. In addition, RID
estimates that each year in the United States, approximately two
million patients contract infections while in hospitals and, of
those, an estimated 100,000 die as a result. According to data
from RID in 2005, post-surgical wound infections more than
double a patient’s hospital costs, and patients with Staph
infections more than triple the average hospital costs. Surgical
site infections account for approximately 500,000 hospital
acquired infections in the United States each year, according to
the CDC.
Staphylococcus aureus, or Staph, is one of the most
common hospital acquired infections. One of the deadliest forms
of Staph infection is MRSA. According to data from the CDC, in
2003, 57% of the Staph infections reported were MRSA, up
from 22% in 1995 and 2% in 1974. Patients who do survive MRSA
often spend months in the hospital and endure repeated surgeries
to remove infected tissue.
When infection is present in a wound, standard treatments can
include cleansing, debridement and systemic antibiotics. Many
cleansing agents can harm tissue, causing irritation and
sensitization and impeding the wound healing process. Some forms
of debridement may increase scar tissue and complicate skin
grafting. Systemic antibiotics may be ineffective if the
patient’s metabolic state is compromised. Additionally, the
effectiveness of oral or systemic antibiotics in diabetic foot
ulcer patients may be diminished due to the patient’s poor
circulation, limiting delivery of the antibiotics to the wound
site.
Because there is a risk of infection with many surgical
procedures, clinicians perform several procedures before and
after surgery designed to prevent infection. Pre-operative
procedures generally involve preparing the surgical site with an
anti-bacterial agent, such as Betadine. Post-operative
procedures can include an anti-infective irrigation, a
therapeutic body cavity cleansing and the use of systemic
antibiotics.
Wound
Healing and Closure
Wound healing is a cascade process comprised of inflammation,
proliferation and maturation. The first stage of the wound
healing process is the inflammatory phase, which is associated
with swelling, redness and heat, and involves the migration of
healthy cells to the wound bed. Removing dead tissue or debris
from the wound prepares the wound bed for regeneration of new
tissue. The second phase is the proliferative phase, which
involves collagen and blood vessel formation and tissue growth.
The final phase, maturation, occurs as the wound begins to take
on its permanent form as collagen is reconstituted, forming new
skin. None of these phases, however, will progress normally in
the presence of infection.
Advanced
Technologies
Techniques and devices have been developed to treat complex and
hard-to-treat wounds, ranging from specialized devices to
antimicrobial dressings. Negative pressure wound therapy, high
pressure oxygen chambers and localized devices, sophisticated
water-based tissue removal devices, oxygenated mist devices and
tissue engineered skin substitutes are some of the most advanced
devices available to the wound care specialist. Although
relatively effective, many of these treatments have limitations
or drawbacks in that they cannot be used on certain types of
wounds or are expensive and complex to use. Despite these
advanced technologies, treatment of challenging wounds continues
to be multi-pronged, with a number of associated therapies
employed in an attempt to achieve wound closure.
Market
Opportunity — Key Limitations of Existing
Treatments
Commonly used topical antiseptics and antibiotics have
limitations and side effects that may constrain their usage. For
example:
|
|
|
| |
•
|
many antiseptics, including Betadine, hydrogen peroxide and
Dakin’s solution, are toxic, can destroy human cells and
tissue, may cause allergic reactions and can impede the wound
healing process;
|
| |
| |
•
|
silver-based products are expensive and require precise dosage
and close monitoring by trained medical staff to minimize the
potential for tissue toxicity allergic reactions and bacterial
resistance; and
|
6
|
|
|
| |
•
|
the increase in antibiotic resistant bacterial strains, such as
MRSA and VRE, have compromised the effectiveness of some widely
used topical antibiotics including Neosporin and Bacitracin.
|
| |
| |
•
|
Oral and systemic antibiotics often are not effective in
treating topical infections and can cause serious side effects.
|
Our
Solution
We believe Microcyn has potential advantages over current
methods of care in the treatment of chronic and acute wounds,
including the following:
|
|
|
| |
•
|
Wound Care Solution. Our 510(k) product is
cleared as a medical device for sale in the United States in
wound cleaning, or debridement, lubricating, moistening and
dressing. Although we do not have the necessary regulatory
approvals to market Microcyn in the United States as a drug,
laboratory testing and physician clinical studies further
suggest that our 510(k) Microcyn product may be effective
against a wide range of bacteria that causes infection in a
variety of acute and chronic wounds. In addition, because of its
mechanism of action, we believe Microcyn does not target
specific strains of bacteria, the practice of which has been
shown to promote the development of resistant bacteria. In
physician clinical studies, our 510(k) Microcyn product has been
used in conjunction with other wound care therapeutic products.
Data from these studies suggest that patients generally
experienced less pain, improved mobility and physical activity
levels and better quality of life.
|
| |
| |
•
|
Non-irritating. Our 510(k) product label
states that our 510(k) product, which is based on our Microcyn
technology, is non-irritating and non-sensitizing to the skin
and eyes. Throughout all our clinical trials and physician
clinical studies to date and since our first commercial sale of
Microcyn in Mexico in 2004, we have received no reports of
serious adverse events related to the use of Microcyn products.
|
| |
| |
•
|
Ease of Use. Our 510(k) product label states
that our 510(k) product requires no special handling
precautions. Our products require no preparation before use or
at time of disposal, and caregivers can use our products without
significant training. In addition, Microcyn can be stored at
room temperature. Unlike other super-oxidized water solutions,
which are typically stable for not more than 48 hours, our
laboratory tests show that Microcyn has a shelf life ranging
from one to two years depending on the size and type of
packaging. Our products are also designed to be complementary to
most advanced technologies to treat serious wounds, such as
negative pressure wound therapy, jet lavage and
tissue-engineered skin substitutes.
|
| |
| |
•
|
Cost-Effectiveness. The treatment of many
wounds requires extended hospitalization and care, including the
use of expensive systemic antibiotics. Infection prolongs the
healing time and necessitates increased use of systemic
antibiotics. We believe that Microcyn has the potential to help
treat infection, accelerate healing time and, in certain cases,
may help reduce the need for systemic antibiotics, reduce the
need for amputation and lead to earlier hospital discharge,
thereby lowering overall patient cost.
|
Our
Strategy
Our goal is to become a worldwide leader in anti-infectives in
treating wounds. We also intend to leverage our expertise in
wound care into additional market opportunities. The key
elements of our strategy include the following:
Obtain
drug regulatory approvals in the United States
We intend to seek additional regulatory clearances and
approvals, which we believe will allow us to accelerate adoption
of our products by wound care specialists worldwide. We have
initiated a Phase II trial which is designed to evaluate
the effectiveness of Microcyn in mildly infected diabetic foot
ulcers with endpoints of clinical cure and improvement in signs
and symptoms of infection supported by microbiological response.
We expect to use more than 10 clinical sites with a target of
enrolling 60 patients, using Microcyn alone, Microcyn plus
an oral antibiotic, and saline plus an oral antibiotic. We
expect to announce the results of our Phase II trial in
autumn of 2007. A well known contract research organization is
coordinating, monitoring and documenting results of this trial.
Following
7
the completion of this trial and a review meeting with the FDA,
we intend to initiate two Phase III trials, enrolling
patients with infected diabetic foot ulcers. We anticipate that
Phase III trials will start in early 2008 and will last
about 12 to 18 months. Results from these Phase II and
Phase III clinical trials are intended to provide the
clinical basis for submission to the FDA of an NDA for the
treatment of infected diabetic foot ulcers.
Drive
adoption of Microcyn as the standard of care in the wound care
market to help prevent and treat infection
We believe our products are well positioned to become the
standard of care in helping to treat infections. We seek to
drive adoption of Microcyn as the standard of care in the wound
care market by establishing strong scientific, evidence-based
rationale for its use. We intend to continue to maintain a
marketing presence in key medical communities throughout the
world through targeted direct marketing, publication in
scientific journals, and sponsorships of physician presentations
at medical conferences and seminars.
Develop
strategic collaborations and distribution in the acute and
chronic wound care market
Outside the United States and Mexico, we intend to pursue
strategic relationships with respect to both sales, marketing
and distribution. To accelerate adoption of our products, we may
enter into strategic relationships with healthcare companies
that have product lines, a sales force and distribution channels
that are complementary to ours. We believe collaborations allow
us to leverage our resources and technology. We intend to pursue
access to these markets through strategic partnerships. We have
engaged an investment banker to assist us in identifying
appropriate partners for development and commercialization of
our products. These relationships may take the form of
co-development, co-promotion, co-marketing or distribution
agreements. For instance, in India we sell through Alkem
Laboratories, the
6th largest
pharmaceutical company in India. This year is the first full
year of the product launch of Microcyn in India. In China, we
recently signed a distribution agreement with China Bao Tai,
which intends to distribute Microcyn to hospitals, doctors and
clinics through Sinopharm, the largest pharmaceutical company in
China, and to independent and retail pharmacies through Lianhua
Supermarkets, after required regulatory approval in China is
obtained.
We currently make Microcyn available under our 510(k) clearances
in the United States primarily through our website, one national
distributor and several regional distributors. We plan for a
more aggressive commercialization and product launch in the
event we obtain drug approval from the FDA. After filing the NDA
with the FDA, we may hire a direct sales force or form a
strategic collaboration with a company that already has an
existing sales force to address the US market.
Develop
strategic partnerships in numerous indications outside the wound
care market
We believe our products have potential applications in several
other large markets, including respiratory, ophthalmology,
dermatology, dental and veterinary markets. We intend to pursue
access to these markets through strategic partnerships.
Microcyn
Platform Technology
Mechanism
of Action
We believe Microcyn’s ability to treat and prevent
infection is based on its uniquely engineered chemistry. As a
result of our proprietary manufacturing process, Microcyn is a
proprietary oxychlorine small molecule formulation that, among
other things, interacts with and inactivates surface proteins on
cell walls and membranes of microorganisms and viruses. The
function of these proteins are varied and play significant roles
in cell communication, nutrient and waste transport and other
required functions for cell viability. Once Microcyn surrounds
single cell microorganisms, it damages these proteins, causing
the cell membrane to rupture, leading to cell death, which we
believe is caused by increased membrane permeability and induced
osmotic pressure imbalance. We continue to study the exact
mechanisms by which protein and structural components of the
bacterial cell walls and membranes, and the protein shell that
surrounds a virus, are affected by Microcyn. This destruction of
the cell appears to occur through a fundamentally different
process than that which occurs as a result of contact with a
bleach-based solution because experiments have demonstrated that
Microcyn kills bleach-resistant bacteria. However, we believe
the
8
solution remains non-irritating to human tissues because human
cells have unique protective mechanisms, are interlocked, and
prevent Microcyn from targeting and surrounding single cells
topically on the body. Our laboratory tests suggest that our
solution does not penetrate and kill multi-cellular organisms
and does not damage or affect human DNA.
In laboratory tests, Microcyn has been shown to destroy certain
biofilms. A biofilm is a complex cluster of microorganisms or
bacteria marked by the formation of a protective shell, allowing
the bacteria to collect and proliferate. It is estimated that
over 65% of microbial infections in the body involve bacteria
growing as a biofilm. Bacteria living in a biofilm typically
have significantly different properties from free-floating
bacteria of the same species. One result of this film
environment is increased resistance to antibiotics and to the
body’s immune system. In chronic wounds, biofilms interfere
with the normal healing process and halt or slow wound closure.
In our laboratory studies, Microcyn was shown to destroy two
common biofilms after five minutes of exposure.
In a recently published case study, Microcyn was shown to
significantly increases the dilation of capillaries in wounds as
indicated by higher levels of oxygen at a wound site after the
application of our product.
It is widely accepted that reducing inflammation surrounding an
injury or wound is beneficial to wound healing. Our independent
laboratory research suggests that Microcyn may inhibit certain
inflammatory responses from allergy-producing, or mast cells.
These reactions are critical components of the body’s
natural inflammatory response to injury or wounds. Our
laboratory research suggests that Microcyn’s interference
with these cells is selective to only the inflammatory response
and does not interfere with other functions of these cells.
Additionally, physician clinical studies suggest that Microcyn
only inhibits this inflammatory activity in tissue that is
directly exposed to the solution.
Microcyn has demonstrated antimicrobial activity against
numerous bacterial, viral and fungal pathogens, including
antibiotic-resistant strains, as evidenced by passing results in
numerous standardized laboratory microbiology tests conducted on
our 510(k) product by a variety of certified independent testing
laboratories. Some of the pathogens against which Microcyn has
demonstrated antimicrobial activity are listed below:
| |
|
|
|
|
|
|
|
Pathogen
|
|
|
|
|
|
|
|
|
Antibiotic-Resistant
Bacteria
|
|
|
|
|
|
|
|
Vancomycin Resistant
Enterococcus faecalis (VRE)
|
|
|
|
|
|
|
|
Methicillin resistant
Staphylococcus aureus (MRSA)
|
|
|
|
|
|
|
|
Other Bacteria
|
|
|
|
|
|
|
|
Acinetobacter
baumanii
|
|
|
|
|
|
|
|
Aspergillus niger
|
|
|
|
|
|
|
|
Clostridium difficile
|
|
|
|
|
|
|
|
Escherichia coli
|
|
|
|
|
|
|
|
Escherichia coli
O157:H7
|
|
|
|
|
|
|
|
Mycobacterium bovis
|
|
|
|
|
|
|
|
Pseudomonas
aeruginosa
|
|
|
|
|
|
|
|
Salmonella typhi
|
|
|
|
|
|
|
|
Viruses
|
|
|
|
|
|
|
|
Human Coronavirus
|
|
|
|
|
|
|
|
Human Immunodeficiency Virus Type
1 — HIV
|
|
|
|
|
|
|
|
Influenza A
|
|
|
|
|
|
|
|
Rhinovirus Type 37
|
|
|
|
|
|
|
|
Fungi
|
|
|
|
|
|
|
|
Candida albicans
|
|
|
|
|
|
|
|
Trichophyton
mentagrophytes
|
|
|
|
|
|
|
In addition to the above mentioned independent laboratory
microbiology tests, a study was completed and published in the
Journal of Hospital Infection in 2005, which was co-authored by
our Director of Medical Affairs,
9
Andres Gutiérrez, M.D., Ph.D., that showed that
Microcyn exerts a wide range of antimicrobial activity
(Landa-Solis, González-Espinosa D, Guzman B, Snyder M,
Reyes-Terán G, Torres K and Gutiérrez AA. Microcyn: a
novel super-oxidized water with neutral pH and disinfectant
activity. J Hosp Infect (UK) 61:
291-299).
Current
Regulatory Approvals and Clearances
All our current products are based on our Microcyn platform
technology. We are able to modify the chemistry of Microcyn by
changing the oxidation-reduction potential, pH-level and
concentrations of specific ions or chemicals, which allows us to
manufacture a variety of solutions, each specifically designed
for maximum efficacy and safety by indication. The indications
for our products vary from country to country due to different
regulatory requirements and standards from jurisdiction to
jurisdiction. The indications below are summaries of the
indications approved by the regulatory authority or authorities
in the listed jurisdiction. The similarly named products have
similar formulations; however, they may not have identical
specifications due to varying requirements in different
jurisdictions’ regulatory agencies. The following is a list
of the regulatory approvals and clearances that Microcyn-based
products have received for our most significant or potentially
significant markets:
| |
|
|
|
|
|
|
|
|
|
|
|
Approval or
|
|
Year of Approval
|
|
|
|
|
Region
|
|
Clearance Type
|
|
or Clearance
|
|
|
Summary Indication
|
|
|
|
United States
|
|
510(k)
|
|
|
2005
|
|
|
Moistening and lubricating
absorbent wound dressings for traumatic wounds.
|
|
|
|
510(k)
|
|
|
2005
|
|
|
Moistening and debriding acute and
chronic dermal lesions.
|
|
|
|
510(k)
|
|
|
2006
|
|
|
Moistening absorbent wound
dressings and cleaning minor cuts.
|
|
European Union
|
|
CE Mark
|
|
|
2004
|
|
|
Debriding, irrigating and
moistening acute and chronic wounds in comprehensive wound
treatment by reducing microbial load and creating moist
environment.
|
|
Mexico
|
|
Product Registration
|
|
|
2004
|
|
|
Antiseptic treatment of wounds and
infected areas.
|
|
|
|
Product Registration
|
|
|
2003
|
|
|
Antiseptic disinfection solution
for high level disinfection of medical instruments, and/or
equipment and clean-rooms, areas of medical instruments,
equipment and clean room areas.
|
|
Canada
|
|
Class II Medical Device
|
|
|
2004
|
|
|
Moistening, irrigating, cleansing
and debriding acute and chronic dermal lesions, diabetic ulcers
and post-surgical wounds.
|
|
India(1)
|
|
Drug License
|
|
|
2006
|
|
|
Cleaning and debriding in wound
management.
|
|
|
|
|
(1) |
|
Drug license held by Indian distributor as required by Indian
law. |
Clinical
Trials
We have initiated a trial, which is designed to evaluate the
effectiveness of Microcyn in mildly infected diabetic foot
ulcers with endpoints of clinical cure and improvement in signs
and symptoms of infection supported by microbiological response.
We expect to use more than 10 clinical sites with a target of
enrolling 60 patients, using Microcyn alone, Microcyn plus
an oral antibiotic, and saline plus an oral antibiotic. We
expect to announce the results of our Phase II trial in
autumn of 2007. A well known contract research organization is
coordinating,
10
monitoring and documenting results of our Phase II trial.
Following the completion of this study and a review meeting with
the FDA, we intend to initiate two Phase III trials. We
anticipate that Phase III trials will start in early 2008
and will last about 12 to 18 months. These Phase III
clinical trials are intended to provide the clinical basis for
submission to the FDA of an NDA for the treatment of infected
diabetic foot ulcers.
Physician
Clinical Studies
In addition to the trials mentioned above, several physicians
and scientists have conducted twenty-one clinical evaluations of
Microcyn generating data suggesting that our 510(k) Microcyn
product is non-irritating to healthy tissue, reduces microbial
load, shortens treatment time and may have the potential to
reduce costs to healthcare providers and patients. We have
sponsored the majority of physicians performing these studies by
supplying Microcyn, unrestricted research grants, paying
expenses or providing honoraria. In some cases, the physicians
who performed these studies also hold equity in our company. The
studies were performed in the United States, Europe, India and
Mexico, and used various endpoints, methods and controls (for
example, saline, antiseptics and antibiotics). These studies
were not intended to be rigorously designed or controlled
clinical trials and, as such, did not have all of the controls
required for clinical trials used to support an NDA submission
to the FDA in that they did not include blinding, randomization,
predefined clinical endpoints, use of placebo and active control
groups or U.S. good clinical practice requirements.
In many cases the physicians who led these studies have
published articles on their studies and results. The following
table lists a selection of articles and publications from
physicians who have completed studies on the use of Microcyn for
wound care and wound irrigation.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
Physician
|
|
Country
|
|
Patients
|
|
|
Publication
|
|
|
|
David E. Allie, M.D.(1)
|
|
U.S.
|
|
|
40
|
|
|
Allie D. Super-Oxidized Dermacyn
in Lower-Extremity Wounds. Wounds, 2006, Jan (Suppl), 3-6
|
|
Tom Wolvos, M.D.(2)
|
|
U.S.
|
|
|
26
|
|
|
Wolvos TA. Advanced Wound Care
with Stable, Super-Oxidized Water. A look at how combination
therapy can optimize wound healing. Wounds, 2006, Jan
(Suppl), 11-13
|
|
Cheryl Bongiovanni, Ph.D.(3)
|
|
U.S.
|
|
|
8
|
|
|
Bongiovanni CM. Superoxidized
Water Improves Wound Care Outcomes in Diabetic Patients.
Diabetic Microvascular Complications Today, 2006,
May-Jun: 11-14
|
|
|
|
|
|
|
3
|
|
|
Biogiovanni CM. Nonsurgical
Management of Chronic Wounds in Patients with Diabetes.
Journal of Vascular Ultrasound, 2006, 30: 215-218,
|
|
Luca Dalla Paola, M.D.(4)
|
|
Italy
|
|
|
218
|
|
|
Dalla Paola L, Brocco E, Senesi,
A, Merico M, De Vido D, Assaloni R, DaRos R. Super-Oxidized
Solution (SOS) Therapy for Infected Diabetic Foot Ulcers.
Wounds, 2006, vol. 18: 262-270
|
|
|
|
|
|
|
|
|
|
Dalla Paola, L. Treating diabetic
foot ulcers with super-oxidized water. Wounds, 2006, Jan
(Suppl), 14-16
|
|
Alberto Piagessi, M.D.(5)
|
|
Italy
|
|
|
33
|
|
|
Clinical Outcomes of Wide
Postsurgical Lesions in the Infected Diabetic Foot Managed With
2 Different Local Treatment Tegimes Compared Using a
Quasi-Experimental Study Design: A Preliminary Communication.
Int. J.Lower Extremity Wounds, 2007 6: 22-27.
|
11
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
Physician
|
|
Country
|
|
Patients
|
|
|
Publication
|
|
|
|
Ariel Miranda, M.D.(5)
|
|
Mexico
|
|
|
64
|
|
|
Miranda-Altamirano A. Reducing
Bacterial Infectious Complications from Burn Wounds. A look at
the use of Oculus Microcyn60 to treat wounds in Mexico.
Wounds, 2006, 18 (Suppl), 17-19
|
|
Lenka Veverkova, M.D.(3)
|
|
Czech Republic
|
|
|
27
|
|
|
Veverkova L, Jedlicka V,
Vesely M, Tejkalova R, Zabranska S, Capov I, Votava M.
Methicilin-resistent Staphylococcus
aureus — problem in health care. J Wound
Healing 2005, 2:201-202.
|
|
Elia Ricci M.D.(6)
|
|
Italy
|
|
|
40
|
|
|
Ricci E, Astolfi S, Cassino R.
Clinical results about an antimicrobial solution (Dermacyn Wound
Care) in the treatment of infected chronic wounds.
17th Conference. EWMA Meeting 2005. Glasgow, UK. May 2-4,
2007. In preparation for publication.
|
|
Steenvoorde, P.M.D, Van
Doorn, L.P., M.A.,
Jacobi, C.E, PhD &
Oskam, J., M.D., PhD.(3)
|
|
Netherlands
|
|
|
10
|
|
|
An unexpected effect of Dermacyn
on infected leg ulcers, J Wound Care 2007, 16: 60-61.
|
Notes
|
|
|
|
(1) |
|
indicates that the physician is an investor and was a member of
our Medical and Business Advisory Board which the Company
dissolved in April 2007, a paid consultant and received research
grants, expense payments, honorarium and Microcyn to complete
the study |
| |
|
(2) |
|
indicates that the physician was a paid consultant, received
expenses in connection with corporate development and licensing
evaluations and is a warrant holder |
| |
|
(3) |
|
indicates that the doctor received Microcyn to complete the study |
| |
|
(4) |
|
indicates that the physician is a paid consultant, was a member
of our Medical and Business Advisory Board, which the Company
dissolved in April 2007, and received expense payments and
Microcyn to complete the study |
| |
|
(5) |
|
indicates that the physician received payments, expense payments
and Microcyn to complete the study |
| |
|
(6) |
|
indicates that the physician received reimbursement of travel
expenses and received Microcyn to complete the study |
There are also several ongoing and planned physician clinical
studies in the United States, Europe and India to assess
Microcyn’s effectiveness in preventing and treating
infections in wounds and are summarized in the table below. For
example, we are supporting a study by Dr. David Armstrong
of the Scholl College of Podiatric Medicine in Chicago, Illinois
and Dr. Andrew Boulton, Head of the Manchester Diabetes
Center at the Manchester Royal Infirmary in the United Kingdom.
Drs. Armstrong and Boulton’s study is a study of
diabetic foot ulcers using the VersaJet, an aggressive
debridement system, in two groups of ten patients each, one
utilizing Microcyn and the other utilizing saline. The endpoints
are microbial load reduction and time to complete wound healing.
Other studies in diabetic foot infections are being conducted by
Dr Alberto Piaggesi, Director of the UO Mallatie del Metabolismo
e Diabetologia, Universitaria Pisana, Italy and Dr Robert
Frykberg, Director of Podiatry Medicine at the Carl T Hayden,
Veterans Administration Medical Center in Arizona. Dr Piaggesi
is investigating the clinical and microbiological response to
Dermacyn, with particular attention to the microscopic
anatomical changes in diseased tissue in the formation of new
blood vessels following therapy. Dr Frykberg is analyzing the
effect of Microcyn on MRSA colonized ulcers which is becoming a
public threat due to the frequency of resistance to antibiotics.
Cheryl Bongiovanni, Ph.D., Director of the Lake Wound
Clinics in Lakeview, Oregon, is conducting case report studies
focusing on blood vessel dilation effects of Microcyn and on the
potential cost savings from the use of Microcyn in treating a
variety of wounds. Dr. Matthew Regulski, who practices at
the Ocean County Foot &
12
Ankle Surgical Associates, in New Jersey, is focusing on the
combination of Microcyn and skin substitutes. Finally, Dr Amar
Pal Suri from New Dehli is investigating the effects of Microcyn
on wound healing rates with patient follow up over several
months. We provided each of these doctors with Microcyn and may
pay their expenses, including travel, hotels and meals, to
attend medical conferences to present their findings. We pay
Drs. Frykberg and Regulski an educational grant in
connection with their studies.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of
|
|
|
|
|
Physician
|
|
Country
|
|
Patients
|
|
|
Publication
|
|
|
|
David Armstrong, D.P.M., PhD.(1)
|
|
U.S.
|
|
|
10
|
|
|
A prospective, multi center,
randomized, single-masked, controlled, clinical investigation of
Microcyn as a replacement solution with Versajet Jet lavage
system
|
|
Andrew Boulton M.D.(1)
|
|
U.K.
|
|
|
10
|
|
|
A prospective, multi center,
randomized, single-masked, controlled, clinical investigation of
Microcyn as are placement solution with Versajet Jet lavage
system
|
|
Alberto Piaggesi M.D.:(1)
|
|
Italy
|
|
|
40
|
|
|
Efficacy and safety of using a new
local antiseptic in managing post-surgical lesions of the
diabetic foot — a prospective, randomized, open
clinical investigation
|
|
Robert Frykberg, D.P.M., M.P.H.(2)
|
|
US
|
|
|
30
|
|
|
A prospective clinical evaluation
of ermacyn in wounds of the lower extremity contaminated,
colonized or critically-colonized with or without MRSA
|
|
Matthew Regulski D.P.M.(2)
|
|
US
|
|
|
20
|
|
|
A prospective, single-center,
open-labeled, saline controlled trial to evaluate the efficacy
and compatibility of
Dermacyn®
wound care with
OASIS®
Wound Matrix for the treatment of non-infected, graft ready
venous ulcers, post-surgical, and post-traumatic wounds
|
|
Amar Pal Singh Suri D.P.M.(1)
|
|
India
|
|
|
100
|
|
|
Role of neutral pH, super-oxidized
solution in the healing of diabetic foot ulcers.
|
Notes
|
|
|
|
(1) |
|
indicates that the physician received payments, expense payments
and Microcyn to complete the study |
| |
|
(2) |
|
indicates that the physician received an educational grant |
13
In addition to the above articles and publications, several
additional papers on the basic science of the technology have
been published or have been submitted for peer review and
publication, including:
| |
|
|
|
|
|
Researchers
|
|
Country
|
|
Publication
|
|
|
|
Landa-Solis,
González-Espinosa
D., Guzman B, Snyder M,
Reyes-Terán G., Torres K. and
Gutiérrez A.A.(1)
|
|
México
|
|
Microcyntm
a novel super-oxidized water with neutral pH and disinfectant
activity. J Hosp Infect (UK) 61: 291-299.
|
|
Gutiérrez, A.A.(1)
|
|
US
|
|
The science behind stable,
super-oxidized water. Exploring the various applications of
super-oxidized solutions. Wounds. 18 (Suppl), 7-10.
|
|
Dalla Paola L. and Faglia E.(2)
|
|
Italy
|
|
Treatment of diabetic foot ulcer:
an overview. Strategies for clinical approach. Current
Diabetes Reviews, 2006, 2, 431-447 431
|
|
González-Espinosa D.,
Pérez-Romano L., Guzman Soriano
B., Arias E., Bongiovanni, C.M.
& Gutiérrez A.A.(1),(3)
|
|
Mexico US
|
|
Effects of neutral super-oxidized
water on human dermal fibroblasts in vitro. International
Wound Journal, 2007. In press.
|
|
Medina-Tamayo J., Balleza-Tapia
H., López, X., Cid, M.E.,
González-Espinosa, D. Gutiérrez
A.A.., and González-Espinosa C.(1)
|
|
Mexico US
|
|
Super-oxidized water inhibits
IgE-antigen- induced degranulation and cytokine release in mast
cells. International Immunophamacology 2007. In press.
|
|
Zahumensky E.
|
|
Czech Republic
|
|
Infections and diabetic foot
syndrome in field practice. Vnitr Lek. 2006;52:411-416.
|
|
Rose R., Setlow B., Monroe A.,
Mallozzi M., Driks A., Setlow P.
|
|
US
|
|
Comparison of the properties of
Bacillus subtilis spores made in liquid or on agar plates.
Submitted 2007.
|
|
Paul M., Setlow B. and Setlow P.
|
|
US
|
|
The killing of spores of Bacillus
subtilis by
Microcyntm,
a stable superoxided water. Submitted 2007.
|
|
Sauer K, Vazquez G., Thatcher
E., Northey R. and
Gutierrez A.A.(1),(4),(5)
|
|
US
|
|
Neutral super-oxidized solution is
effective in killing P. aeruginosa biofilms. Submitted
2007.
|
Notes
|
|
|
|
(1) |
|
Dr. Gutierrez is our Director of Medical Affairs and
conducted the study during his employment by the Company |
| |
|
(2) |
|
Dr. Dalla Paola was a member of our Medical and Business
Advisory Board, which the Company dissolved in April 2007, and
received expense payments and Microcyn to complete the study |
| |
|
(3) |
|
Dr. Bongiovanni received Microcyn to complete the study |
| |
|
(4) |
|
Dr. Thatcher is a full-time consultant to us, holds shares
of our stock, previously served on our board of directors, and
received Microcyn to complete the study |
| |
|
(5) |
|
Dr. Northey is our Director of Research &
Development and conducted the study during his employment by the
Company |
Sales and
Marketing
Our products are purchased by hospitals, physicians, nurses and
other healthcare practitioners who are the primary caregivers to
patients being treated for acute or chronic wounds, as well as
those patients undergoing surgical procedures. In the United
States, we make Microcyn available under 510(k) clearances
primarily through our website, one national distributor and
several regional distributors. We plan for a more aggressive
commercialization and product launch in the event we receive
drug approval from the FDA.
14
In Europe, we have arrangements with distributors in Germany,
Italy, and the Czech Republic. We are actively pursuing
strategic alliances with potential partners, that already have
significant sales, marketing and distribution capabilities to
hospitals and pharmacies throughout Europe. It is our intent to
use the partners’ capabilities to sell and distribute
Microcyn in Europe.
In Mexico, we market our products through our established
distribution network and direct sales organization. We have a
dedicated contract sales force, including salespeople, nurses
and clinical support staff responsible for selling Microcyn to
private and public hospitals and to retail pharmacies.
Throughout the rest of the world, we intend to use strategic
partners and distributors, that have a significant sales,
marketing and distribution presence in their respective
countries. We have established partners and distribution
channels for our wound care products in China, India,
Bangladesh, Pakistan, Singapore, United Arab Emirates and Saudi
Arabia.
In December 2005, we entered into an agreement with Alkem
Laboratories, a large pharmaceutical company in India. We
commenced sales to Alkem Laboratories in April 2006. Under the
terms of this agreement, Alkem has exclusive rights to market,
distribute and sell our Microcyn-based products in the Republic
of India and the Kingdom of Nepal. During the term of this
agreement, Alkem is entitled to use our patents, trade secrets,
trademarks and other intellectual property rights as to our
Microcyn-based products solely in connection with our products.
However, we will remain the owner of and reserve such patents,
trade secrets, trademarks and other intellectual property
rights. In the event we fail to timely deliver the ordered
quantities, we will be subject to certain penalties. In
addition, if either party fails to fulfill their respective
obligations under the agreement for a period of 180 days,
which is not remediated within 30 days of receiving notice,
the other party may terminate the agreement. The agreement has a
five year term and may be renewed after its initial term for
such additional term as the parties agree to in writing.
In April 2007, we entered into an exclusive distribution
agreement with China Bao Tai Investment Company, Ltd., or China
Bao Tai, for the sale of Microcyn wound care solution in China,
Hong Kong, Macau and Taiwan. China Bao Tai intends to distribute
and sell Microcyn to hospitals and pharmacies through Sinopharm,
the largest pharmaceutical group in China, and through Lianhua
Supermarkets for supermarket distribution. Lianhua Supermarkets
operates 3,609 outlets spanning 21 provinces. This agreement
provides for minimum purchases of $12 million over a five
year period, with the minimum purchases heavily weighted to the
latter years of sales. If those minimums are not met, we are not
bound by our exclusivity agreement. Sales of Microcyn wound care
solution under this agreement will commence after China Bao Tai
has obtained the required regulatory approval for distribution
of the products in the territories, which it expects to receive
in late 2009.
Other
Market Opportunities
We are searching for strategic partnerships in addition to wound
care applications in markets where Microcyn technology has
competitive advantages over antibiotics in numerous medical
indications outside of the acute and surgical wound market. Some
of these market opportunities include:
Respiratory
Our nasal product candidate is an anti-microbial solution
designed to be self-administered into a patient’s nasal
cavity for the treatment of chronic rhinosinusitis, or
inflammation of the nasal sinuses. In animal studies, Microcyn
has been shown to kill the bacteria that causes rhinosinusitis.
We have conducted pre-clinical animal studies that suggest the
efficacy and safety of this product candidate.
Rhinosinusitis affects an estimated 35 million people in
the United States. There is no FDA-approved therapy for chronic
rhinosinusitis. Most treatment methods have focused on the
symptoms of the disease and include the use of antibiotics,
antihistamines, corticosteroids and sinus surgery.
15
Dermatology
We believe that our Microcyn technology can be used to develop
products to treat various fungal and bacterial skin infections.
Laboratory and clinical test data support that our technology
may be effective in treating these bacterial and fungal
infections.
In February 2007, we entered into an exclusive agreement with
Dancohr Corporation B.V., a manufacturer and wholesaler of
cosmetics, to distribute Courtin oxychlorine solution to salons
in various European countries for cleaning hands and feet during
pedicures and manicures. This agreement provides for minimum
purchases of $10 million over a five year period, with
minimum purchases of €30,000 in 2007. If minimum purchases
are not met, we may terminate the agreement.
Dental
and Oral Care
We believe that our Microcyn technology may be used both as a
mouthwash and a dental rinse, and that early data from physician
studies support its safe use in oral surgery.
Ophthamology
We believe that our technology may be used to treat and prevent
eye infections such as conjunctivitis. We have conducted
in vitro and animal laboratory testing that suggests that
our product is safe when placed in the eye.
Veterinary
Medicine
Our animal wound care product based on Microcyn technology,
Vetericyn, is available for use in the United States, and
we are seeking to identify a partner with the expertise and
capability to exploit the verterinary market.
Research
and Product Development
The main goals of our research and product development program
are to design, develop and produce products to treat acute and
chronic wounds, and to identify new applications for our
technology. Our research and product development efforts with
our Microcyn-based products are divided into three areas:
science, new product development and engineering.
Our scientists work to continually improve our product
performance by evaluating variations of the formulations and
chemical structures of our products. For example, we are
evaluating alterations to Microcyn to increase the speed at
which it kills certain bacteria and viruses. Significant efforts
are also being directed towards extending our understanding of
the unique chemistry of our products.
The focus of our current development efforts is new
formulations, applications and delivery systems for Microcyn,
including the following:
|
|
|
| |
•
|
Modification of the physical properties of our product to
improve efficiency in unique antimicrobial applications;
|
| |
| |
•
|
Development of new formulations and delivery systems that extend
the stability of the product;
|
| |
| |
•
|
Development of a surgical irrigant to control infections during
and after surgery; and
|
| |
| |
•
|
Alteration of current formulation for sinus treatments.
|
Currently, the main focus of our engineering staff is the
construction of a U.S. Good Manufacturing Practices, or
cGMP, compliant manufacturing system for our drug product. This
entails significant upgrades of our raw material, manufacturing
and bottling system.
Our technology may have application in other non-medical
markets. We intend to pursue opportunities in these markets with
third parties. We plan to increase our research and product
development staff in the future to address market demands
identified in our market research and commercial practice.
16
Manufacturing
We manufacture Microcyn through a proprietary electrolysis
process within a multi-chamber system. We are able to control
the passage of ions through proprietary membranes, yielding
electrolyzed water with only trace amounts of chlorine. This
process is fundamentally different from the processes for
manufacturing hydrogen peroxide and bleach and is the basis for
our technology’s effectiveness and safety. Our
manufacturing process produces very little waste, which is
disposed of as water after a simple non-toxic chemical treatment.
We manufacture our products in Petaluma, California, Sittard,
The Netherlands and Zapopan, Mexico. We have developed an
automated manufacturing process and conduct quality assurance
testing on each production batch in accordance with current
U.S. cGMP. Our facilities are required to meet and maintain
regulatory standards applicable to the manufacture
pharmaceutical and medical device products. Our United States
and Netherlands facilities are certified and comply with cGMP
medical device Quality Systems Regulation, or QSR, and
International Organization for Standardization, or ISO,
guidelines. Our Mexico facility has been approved by the MOH.
Our machines are subjected to a series of tests, which is part
of a validation protocol mandated by cGMP, QSR and ISO
requirements. This validation is designed to ensure that the
final product is consistently manufactured in accordance with
product specifications at all manufacturing sites. Certain
materials and components used in manufacturing our machines are
proprietary to us.
We believe we have a sufficient number of machines to produce an
adequate amount of Microcyn to meet anticipated future
requirements for at least the next two years. As we expand into
new geographic markets, we may establish additional
manufacturing facilities to better serve those new markets.
Intellectual
Property
Our success depends in part on our ability to obtain and
maintain proprietary protection for our product technology and
know-how, to operate without infringing proprietary rights of
others, and to prevent others from infringing our proprietary
rights. We seek to protect our proprietary position by, among
other methods, filing, when possible, U.S. and foreign
patent applications relating to our technology, inventions and
improvements that are important to our business. We also rely on
trade secrets, know-how, continuing technological innovation,
and in-licensing opportunities to develop and maintain our
proprietary position.
As of May 31, 2007, we own one issued U.S. patent, two
issued European patents, 13 pending U.S. patent
applications and 20 foreign pending patent applications
generally relating to super-oxidized water. These applications
include six international PCT applications that have not yet
reached the deadline to file counterpart phase applications. Our
portfolio of issued and pending applications can be divided into
two groups. The first group includes one U.S. issued
patent, two issued European patents, three pending
U.S. patent applications and five foreign patent
applications that relate to early generation super-oxidized
water product, methods of using super-oxidized water, and
aspects of the method and apparatus for manufacturing
super-oxidized water. The second group includes 10 pending
U.S. patent applications and 15 foreign patent applications
that relate to Microcyn, the method and apparatus for
manufacturing Microcyn, and its uses.
In March 2003, we obtained an exclusive license to six issued
Japanese patents and five Japanese published pending patent
applications owned by Coherent Technologies, or Coherent. The
issued Japanese patents and pending Japanese patent applications
relate to an early generation of unstable, super-oxidized water
product and aspects of the method and apparatus for producing
super-oxidized water and will expire between 2011 and 2014. In
June 2006, we received written notice via email from Coherent
advising us that the patent license was terminated, citing
various reasons with which we disagree. Although we do not
believe Coherent has grounds to terminate the license, we may
have to take legal action to preserve our rights under the
license and to enjoin Coherent from breaching its terms. We do
not know whether we would prevail in any such action, which
would be costly and time consuming, and we could lose our rights
under the license, which could have a material adverse impact on
our business opportunities in Japan. In addition, we may have to
defend ourselves against infringement claims from Coherent in
Japan based on their position on termination of the license. We
do not believe the Japanese patents disclose or cover certain
innovations in our products, which we developed independently
and are the subject of our own patent applications. Neither
party has taken any formal legal action in connection with
Coherent assertions. In
17
fact, we maintain an ongoing dialogue with Coherent. To date, we
have not commercialized any products or generated any revenue in
Japan.
Although we work to protect our technology, we cannot assure you
that any patent will issue from currently pending patent
applications or from future patent applications. We also cannot
assure you that the scope of any patent protection will exclude
competitors or provide competitive advantages to us, that any of
our patents will be held valid if subsequently challenged, or
that others will not claim rights in or ownership of our patents
and proprietary rights. Furthermore, we cannot assure you that
others have not developed or will develop similar products,
duplicate any of our products or design around our patents.
We have also filed for trademark protection for marks used with
our Microcyn products in each of the United States, Europe,
Canada, certain countries in Central and South America,
including Mexico and Brazil, and certain countries in Asia,
including Japan, China, the Republic of Korea, India and
Australia. In addition to patents and trademarks, we rely on
trade secret and other intellectual property laws, nondisclosure
agreements and other measures to protect our intellectual
property rights. We believe that in order to have a competitive
advantage, we must develop and maintain the proprietary aspects
of our technologies. We require our employees, consultants and
advisors to execute confidentiality agreements in connection
with their employment, consulting or advisory relationship with
us. We also require our employees, consultants and advisors who
we expect to work on our products to agree to disclose and
assign to us all inventions made in the course of our working
relationship with them, while using our property or which relate
to our business. Despite any measures taken to protect our
intellectual property, unauthorized parties may attempt to copy
aspects of our products or to wrongfully obtain or use
information that we regard as proprietary. For more information,
please see “Risk Factors,” “Our competitive
position depends on our ability to protect our intellectual
property and our proprietary technologies.”
Competition
We believe the principal competitive factors in our target
market include improved patient outcomes, such as time in the
hospital, healing time, adverse events, safety of products, ease
of use, stability, spore killing and cost effectiveness. The
wound care market is highly competitive. We compete with a
number of large, well-established and well-funded companies that
sell a broad range of wound care products, including topical
anti-infectives and antibiotics, as well as some advanced wound
technologies, such as skin substitutes, growth factors and
sophisticated delayed release silver-based dressings.
Our products compete with a variety of products used for wound
cleaning, debriding and moistening, including sterile saline,
and chlorhexadine-based products, and they also compete with a
large number of prescription and over-the-counter products for
the prevention and treatment of infections, including topical
anti-infectives, such as Betadine, silver sulfadiazine, hydrogen
peroxide, Dakin’s solution and hypochlorous acid, and
topical antibiotics, such as Neosporine, Mupirocin and
Bacitracin. Currently, no single anti-infective product
dominates the chronic or acute wound markets because many of the
products have serious limitations or tend to inhibit the wound
healing process.
Our products can also replace the use of sterile saline for
debriding and moistening a dressing as well as for use as a
complementary product with many advanced wound care
technologies, such as the VACTherapy System from Kinetic
Concepts Inc., skin substitute products from Smith &
Nephew, Integra Life Sciences, Life Cell, Organogenesis and
Ortec International, and ultrasound from Celleration. We believe
that Microcyn can enhance the effectiveness of many of these
advanced wound care technologies. Because Microcyn is
competitive with some of the large wound care companies’
products and complementary to others, we may compete with such
companies in some product lines and complement other product
lines.
While many companies are able to produce oxidized water, their
products, unlike ours, typically become unstable after
48 hours, and we believe they have a much higher chlorine
content that may not be suitable for treatment of infections in
wounds. One such company, PuriCore, sells electrolysis machines
used to manufacture brine-based oxidized water primarily as a
sterilant.
Some of our competitors enjoy several competitive advantages,
including:
|
|
|
| |
•
|
significantly greater name recognition;
|
18
|
|
|
| |
•
|
established relationships with healthcare professionals,
patients and third party payors;
|
| |
| |
•
|
established distribution networks;
|
| |
| |
•
|
additional product lines and the ability to offer rebates or
bundle products to offer discounts or incentives;
|
| |
| |
•
|
greater experience in conducting research and development,
manufacturing, obtaining regulatory approval for products and
marketing; and
|
| |
| |
•
|
greater financial and human resources for product development,
sales and marketing and patient support.
|
Government
Regulation
Government authorities in the United States at the federal,
state and local levels and foreign countries extensively
regulate, among other things, the research, development,
testing, manufacture, labeling, promotion, advertising,
distribution, sampling, marketing, and import and export of
pharmaceutical products, biologics and medical devices. All of
our products in development will require regulatory approval by
government agencies prior to commercialization. In particular,
human therapeutic products are subject to rigorous pre- clinical
and clinical trials and other approval procedures of the FDA and
similar regulatory authorities in foreign countries. Various
federal, state, local and foreign statutes and regulations also
govern testing, manufacturing, safety, labeling, storage,
distribution and record-keeping related to such products and
their marketing. The process of obtaining these approvals and
the subsequent process of maintaining substantial compliance
with appropriate federal, state, local, and foreign statutes and
regulations require the expenditure of substantial time and
financial resources. In addition, statutes, rules, regulations
and policies may change and new legislation or regulations may
be issued that could delay such approvals.
Medical
Device Regulation
In 2005, Microcyn received 510(k) clearance as a medical device
for wound cleaning, or debridement, lubricating, moistening and
dressing. Any future product candidates or new applications
using Microcyn that are classified as medical devices will need
approval or clearance by the FDA.
New medical devices, such as Microcyn, are subject to FDA
clearance and extensive regulation under the Federal Food Drug
and Cosmetic Act, or FDCA. Under the FDCA, medical devices are
classified into one of three classes: Class I,
Class II or Class III. The classification of a device
into one of these three classes generally depends on the degree
of risk associated with the medical device and the extent of
control needed to ensure safety and effectiveness.
Class I devices are those for which safety and
effectiveness can be assured by adherence to a set of general
controls. These general controls include compliance with the
applicable portions of the FDA’s Quality System Regulation,
which sets forth good manufacturing practice requirements;
facility registration, device listing and product reporting of
adverse medical events; truthful and non-misleading labeling;
and promotion of the device only for its cleared or approved
intended uses. Class II devices are also subject to these
general controls, and any other special controls as deemed
necessary by the FDA to ensure the safety and effectiveness of
the device. Review and clearance by the FDA for these devices is
typically accomplished through the so-called 510(k) pre-market
notification procedure. When 510(k) clearance is sought, a
sponsor must submit a pre-market notification demonstrating that
the proposed device is substantially equivalent to a legally
marketed Class II device (for example, a device previously
cleared through the 510(k) premarket notification process). If
the FDA agrees that the proposed device is substantially
equivalent to the predicate device, then 510(k) clearance to
market will be granted. After a device receives 510(k)
clearance, any modification that could significantly affect its
safety or effectiveness, or that would constitute a major change
in its intended use, requires a new 510(k) clearance or could
require pre-market approval, or PMA.
Clinical trials are almost always required to support a PMA
application and are sometimes required for a 510(k) pre-market
notification. These trials generally require submission of an
application for an investigational device exemption, or IDE. An
IDE must be supported by pre-clinical data, such as animal and
laboratory testing results, which show that the device is safe
to test in humans and that the study protocols are
scientifically sound. The
19
IDE must be approved in advance by the FDA for a specified
number of patients, unless the product is deemed a
non-significant risk device and is eligible for more abbreviated
investigational device exemption requirements.
Both before and after a medical device is commercially
distributed, manufacturers and marketers of the device have
ongoing responsibilities under FDA regulations. The FDA reviews
design and manufacturing practices, labeling and record keeping,
and manufacturers’ required reports of adverse experiences
and other information to identify potential problems with
marketed medical devices. Device manufacturers are subject to
periodic and unannounced inspection by the FDA for compliance
with the Quality System Regulation, which sets forth the current
good manufacturing practice requirements that govern the methods
used in, and the facilities and controls used for, the design,
manufacture, packaging, servicing, labeling, storage,
installation and distribution of all finished medical devices
intended for human use.
FDA regulations prohibit the advertising and promotion of a
medical device for any use outside the scope of a 510(k)
clearance or PMA approval or for unsupported safety or
effectiveness claims. Although the FDA does not regulate
physicians’ practice of medicine, the FDA does regulate
manufacturer communications with respect to off-label use.
If the FDA finds that a manufacturer has failed to comply with
FDA laws and regulations or that a medical device is ineffective
or poses an unreasonable health risk, it can institute or seek a
wide variety of enforcement actions and remedies, ranging from a
public warning letter to more severe actions such as:
|
|
|
| |
•
|
fines, injunctions and civil penalties;
|
| |
| |
•
|
recall or seizure of products;
|
| |
| |
•
|
operating restrictions, partial suspension or total shutdown of
production;
|
| |
| |
•
|
refusing requests for 510(k) clearance or PMA approval of new
products;
|
| |
| |
•
|
withdrawing 510(k) clearance or PMA approvals already
granted; and
|
| |
| |
•
|
criminal prosecution.
|
The FDA also has the authority to require repair, replacement or
refund of the cost of any medical device.
The FDA also administers certain controls over the export of
medical devices from the United States, as international sales
of medical devices that have not received FDA clearance are
subject to FDA export requirements. Additionally, each foreign
country subjects such medical devices to its own regulatory
requirements. In the European Union, a single regulatory
approval process has been created, and approval is represented
by the CE Mark.
Pharmaceutical
Product Regulation
We have two pharmaceutical product candidates that are regulated
by the FDA and will require approval before we can market or
sell them as drugs. Any future product candidates or new
applications using Microcyn that are classified as drugs will
need approval by the FDA.
In the United States, the FDA regulates drugs under the FDCA and
implementing regulations that are adopted under the FDCA. In the
case of biologics, the FDA regulates such products under the
Public Health Service Act. If we fail to comply with the
applicable requirements under these laws and regulations at any
time during the product development process, approval process,
or after approval, we may become subject to administrative or
judicial sanctions. These sanctions could include the FDA’s
refusal to approve pending applications, withdrawals of
approvals, clinical holds, warning letters, product recalls,
product seizures, total or partial suspension of our operations,
injunctions, fines, civil penalties or criminal prosecution. Any
agency enforcement action could have a material adverse effect
on us. The FDA also administers certain controls over the export
of drugs and biologics from the United States.
|
|
|
| |
•
|
Pre-ClinicalPhase. The pre-clinical phase
involves the discovery, characterization, product formulation
and animal testing necessary to prepare an Investigational New
Drug application, or IND, for submission to the FDA. The IND
must be accepted by the FDA before the drug can be tested in
humans.
|
20
|
|
|
| |
•
|
Clinical Phase. The clinical phase of
development follows a successful IND submission and involves the
activities necessary to demonstrate the safety, tolerability,
efficacy, and dosage of the substance in humans, as well as the
ability to produce the substance in accordance with cGMP
requirements. Data from these activities are compiled in a New
Drug Application, or NDA, or for biologic products a Biologics
License Application, or BLA, for submission to the FDA
requesting approval to market the drug.
|
| |
| |
•
|
Post-Approval Phase. The post-approval phase
follows FDA approval of the NDA or BLA, and involves the
production and continued analytical and clinical monitoring of
the product. The post- approval phase may also involve the
development and regulatory approval of product modifications and
line extensions, including improved dosage forms, of the
approved product, as well as for generic versions of the
approved drug, as the product approaches expiration of patent or
other exclusivity protection.
|
Each of these three phases is discussed further below.
Pre-Clinical Phase. The development of a new
pharmaceutical agent begins with the discovery or synthesis of a
new molecule. These agents are screened for pharmacological
activity using various animal and tissue models, with the goal
of selecting a lead agent for further development. Additional
studies are conducted to confirm pharmacological activity, to
generate safety data, and to evaluate prototype dosage forms for
appropriate release and activity characteristics. Once the
pharmaceutically active molecule is fully characterized, an
initial purity profile of the agent is established. During this
and subsequent stages of development, the agent is analyzed to
confirm the integrity and quality of material produced. In
addition, development and optimization of the initial dosage
forms to be used in clinical trials are completed, together with
analytical models to determine product stability and
degradation. A bulk supply of the active ingredient to support
the necessary dosing in initial clinical trials must be secured.
Upon successful completion of pre-clinical safety and efficacy
studies in animals, an IND submission is prepared and provided
to the FDA for review prior to commencement of human clinical
trials. The IND consists of the initial chemistry, analytical,
formulation, and animal testing data generated during the
pre-clinical phase. The review period for an IND submission is
30 days, after which, if no comments are made by the FDA,
the product candidate can be studied in Phase I clinical trials.
Clinical Phase. Following successful
submission of an IND, the sponsor is permitted to conduct
clinical trials involving the administration of the
investigational product candidate to human subjects under the
supervision of qualified investigators in accordance with good
clinical practice. Clinical trials are conducted under protocols
detailing, among other things, the objectives of the study and
the parameters to be used in assessing the safety and the
efficacy of the drug. Each protocol must be submitted to the FDA
as part of the IND prior to beginning the trial. Each trial must
be reviewed, approved and conducted under the auspices of an
independent Institutional Review Board, and each trial, with
limited exceptions, must include the patient’s informed
consent. Typically, clinical evaluation involves the following
time-consuming and costly three-phase sequential process:
|
|
|
| |
•
|
Phase I. Phase I human clinical trials are
conducted in a limited number of healthy individuals to
determine the drug’s safety and tolerability and include
biological analyses to determine the availability and
metabolization of the active ingredient following
administration. The total number of subjects and patients
included in Phase I clinical trials varies, but is generally in
the range of 20 to 80 people.
|
| |
| |
•
|
Phase II. Phase II clinical trials
involve administering the drug to individuals who suffer from
the target disease or condition to determine the drug’s
potential efficacy and ideal dose. These clinical trials are
typically well controlled, closely monitored, and conducted in a
relatively small number of patients, usually involving no more
than several hundred subjects. These trials require scale up for
manufacture of increasingly larger batches of bulk chemical.
These batches require validation analysis to confirm the
consistent composition of the product.
|
| |
| |
•
|
Phase III. Phase III clinical trials are
performed after preliminary evidence suggesting effectiveness of
a drug has been obtained and safety (toxicity), tolerability,
and an ideal dosing regimen have been established.
Phase III clinical trials are intended to gather additional
information about the effectiveness and safety that is needed to
evaluate the overall benefit-risk relationship of the drug and
to complete the information needed to provide adequate
instructions for the use of the drug. Phase III trials
usually include from several hundred to several thousand
subjects.
|
21
Throughout the clinical phase, samples of the product made in
different batches are tested for stability to establish shelf
life constraints. In addition, large-scale production protocols
and written standard operating procedures for each aspect of
commercial manufacture and testing must be developed.
Phase I, II, and III testing may not be completed
successfully within any specified time period, if at all. The
FDA closely monitors the progress of each of the three phases of
clinical trials that are conducted under an IND and may, at its
discretion, reevaluate, alter, suspend, or terminate the testing
based upon the data accumulated to that point and the FDA’s
assessment of the risk/benefit ratio to the patient. Clinical
investigators, or IRBs, and companies may be subject to
pre-approval, routine, or “for cause” inspections by
the FDA for compliance with Good Clinical Practices, or GCPs,
and FDA regulations governing clinical investigations. The FDA
may suspend or terminate clinical trials, or a clinical
investigator’s participation in a clinical trial, at any
time for various reasons, including a finding that the subjects
or patients are being exposed to an unacceptable health risk.
The FDA can also request additional clinical trials be conducted
as a condition to product approval. Additionally, new government
requirements may be established that could delay or prevent
regulatory approval of our products under development.
Furthermore, institutional review boards, which are independent
entities constituted to protect human subjects in the
institutions in which clinical trials are being conducted, have
the authority to suspend clinical trials in their respective
institutions at any time for a variety of reasons, including
safety issues.
Post-Approval Phase. After approval, we are
still subject to continuing regulation by the FDA, including,
but not limited to, record keeping requirements, submitting
periodic reports to the FDA, reporting of any adverse
experiences with the product, and complying with drug sampling
and distribution requirements. In addition, we are required to
maintain and provide updated safety and efficacy information to
the FDA. We are also required to comply with requirements
concerning advertising and promotional labeling. In that regard,
our advertising and promotional materials must be truthful and
not misleading. We are also prohibited from promoting any
non-FDA approved or “off-label” indications of
products. Failure to comply with those requirements could result
in significant enforcement action by the FDA, including warning
letters, orders to pull the promotional materials, and
substantial fines. Also, quality control and manufacturing
procedures must continue to conform to cGMP after approval.
Drug and biologics manufacturers and their subcontractors are
required to register their facilities and products manufactured
annually with the FDA and certain state agencies and are subject
to periodic routine and unannounced inspections by the FDA to
assess compliance with cGMP regulations. Facilities may also be
subject to inspections by other federal, foreign, state, or
local agencies. In addition, approved biological drug products
may be subject to
lot-by-lot
release testing by the FDA before these products can be
commercially distributed. Accordingly, manufacturers must
continue to expend time, money, and effort in the area of
production and quality control to maintain compliance with cGMP
and other aspects of regulatory compliance. Future FDA
inspections may identify compliance issues at our facilities or
at the facilities that may disrupt production or distribution,
or require substantial resources to correct.
In addition, following FDA approval of a product, discovery of
problems with a product or the failure to comply with
requirements may result in restrictions on a product,
manufacturer, or holder of an approved marketing application,
including withdrawal or recall of the product from the market or
other voluntary or FDA-initiated action that could delay further
marketing. Newly discovered or developed safety or effectiveness
data may require changes to a product’s approved labeling,
including the addition of new warnings and contraindications.
Also, the FDA may require post-market testing and surveillance
to monitor the product’s safety or efficacy, including
additional clinical studies, known as Phase IV trials, to
evaluate long-term effects.
Regulation
of Disinfectants
In October 2004, we obtained EPA authorization, or registration
for the distribution and sale of our Microcyn-based product as a
hospital grade disinfectant. In August 2006, we received a
“show cause” letter from the EPA stating that it was
prepared to file a civil administrative complaint against us for
violation of federal pesticide legislation in connection with
the sale or distribution of a pesticide that did not meet the
label’s efficacy claims. On April 5, 2007, we entered
into a Consent Agreement and Final Order with the EPA allowing
us to amend our EPA registration to a food sanitizer and pay a
$20,800 fine without admitting or denying any wrongdoing.
22
In the United States, the EPA regulates disinfectants as
antimicrobial pesticides under the Federal Insecticide,
Fungicide and Rodenticide Act, or FIFRA, and the implementing
regulations that the EPA has adopted under FIFRA. Before
marketing a disinfectant in the United States, we must satisfy
the EPA’s pesticide registration requirements. That
registration process requires us to demonstrate the
disinfectant’s efficacy and to determine the potential
human and ecological risks associated with use of the
disinfectant. The testing and registration process could be
lengthy and could be expensive. There is no assurance, however,
that we will be able to satisfy all of the pesticide
registration requirements for a particular proposed new
disinfectant product. Once we satisfy the FIFRA registration
requirements for an individual disinfectant, additional FIFRA
regulations will apply to our various business activities,
including marketing, related to that EPA-registered product.
Failure to comply with FIFRA’s requirements could expose us
to various enforcement actions. FIFRA empowers the EPA to seek
administrative or judicial sanctions against those who violate
FIFRA. Among the potential FIFRA penalties are civil
administrative penalties, stop sale orders, cancellation of our
registration, seizures, injunctions and criminal sanctions. If
EPA were to initiate a FIFRA enforcement action against us, it
could have a material adverse effect on us.
Other
Regulation in the United States
Health
Care Coverage and Reimbursement by Third-Party Payors
Commercial success in marketing and selling our products
depends, in part, on the availability of adequate coverage and
reimbursement from third-party health care payors, such as
government and private health insurers and managed care
organizations. Third-party payers are increasingly challenging
the pricing of medical products and services. Government and
private sector initiatives to limit the growth of health care
costs, including price regulation, competitive pricing, and
managed-care arrangements, are continuing in many countries
where we do business, including the United States. These changes
are causing the marketplace to be more cost-conscious and
focused on the delivery of more cost-effective medical products.
Government programs, including Medicare and Medicaid, private
health care insurance companies, and managed-care plans control
costs by limiting coverage and the amount of reimbursement for
particular procedures or treatments. This has created an
increasing level of price sensitivity among customers for our
products. Some third-party payors also require that a favorable
coverage determination be made for new or innovative medical
devices or therapies before they will provide reimbursement of
those medical devices or therapies. Even though a new medical
product may have been cleared or approved for commercial
distribution, we may find limited demand for the product until
adequate coverage and reimbursement have been obtained from
governmental and other third-party payors.
Fraud and
Abuse Laws
In the United States, we are subject to various federal and
state laws pertaining to healthcare fraud and abuse, which,
among other things, prohibit the offer or acceptance of
remuneration intended to induce or in exchange for the purchase
of products or services reimbursed under a federal healthcare
program and the submission of false or fraudulent claims with
the government. These laws include the federal Anti-Kickback
Statute, the False Claim Act and comparable state laws. These
laws regulate the activities of entities involved in the
healthcare industry, such as us, by limiting the kinds of
financial arrangements such entities may have with healthcare
providers who use or recommend the use of medical products
(including for example, sales and marketing programs, advisory
boards and research and educational grants). In addition, in
order to ensure that healthcare entities comply with healthcare
laws, the Office of Inspector General, or OIG, of the
U.S. Department of Health and Human Services recommends
that healthcare entities institute effective compliance
programs. To assist in the development of effective compliance
programs, the OIG has issued model Compliance Program Guidance,
or CPG, materials for a variety of healthcare entities which,
among other things, identify practices to avoid that may
implicate the federal Anti-Kickback Statute and other relevant
laws and describes elements of an effective compliance program.
While compliance with the CPG materials is voluntary, a recent
California law requires pharmaceutical and devices manufacturers
to initiate compliance programs that incorporate the CPG and the
July 2002 Pharmaceuticals Research and Manufacturers of America
Code on Interactions with Healthcare Professionals.
23
Due to the scope and breadth of the provisions of some of these
laws, it is possible that some of our practices might be
challenged by the government under one or more of these laws in
the future. Violations of these laws, which are discussed more
fully below, can lead to civil and criminal penalties, damages,
imprisonment, fines, exclusion from participation in Medicare,
Medicaid and other federal health care programs, and the
curtailment or restructuring of our operations. Any such
violations could have a material adverse effect on our business,
financial condition, results of operations or cash flows.
Anti-Kickback Laws. Our operations are subject
to federal and state anti-kickback laws. The federal
Anti-Kickback Statute prohibits persons from knowingly and
willfully soliciting, receiving, offering or providing
remuneration directly or indirectly to induce either the
referral of an individual for a good or service reimbursed under
a federal healthcare program, or the furnishing, recommending,
or arranging of a good or service, for which payment may be made
under a federal healthcare program, such as Medicare or
Medicaid. The definition of “remuneration” has been
broadly interpreted to include anything of value, including such
items as gifts, discounts, the furnishing of supplies or
equipment, waiver of co-payments, and providing anything at less
than its fair market value. Because the Anti-Kickback Statute
makes illegal a wide variety of common (even beneficial)
business arrangements, the OIG was tasked with issuing
regulations, commonly known as “safe harbors,” that
describe arrangements where the risk of illegal remuneration is
minimal. As long as all of the requirements of a particular safe
harbor are strictly met, the entity engaging in that activity
will not be prosecuted under the federal Anti-Kickback Statute.
The failure of a transaction or arrangement to fit precisely
within one or more safe harbors does not necessarily mean that
it is illegal or that prosecution will be pursued. However,
business arrangements that do not fully satisfy an applicable
safe harbor may result in increased scrutiny by government
enforcement authorities, such as the OIG. Our agreements to pay
compensation to our advisory board members and physicians who
conduct clinical trials or provide other services for us may be
subject to challenge to the extent they do not fall within
relevant safe harbors under state and federal anti-kickback
laws. In addition, many states have adopted laws similar to the
federal Anti-Kickback Statute which apply to the referral of
patients for healthcare services reimbursed by Medicaid, and
some have adopted such laws with respect to private insurance.
Violations of the Anti-Kickback Statute are subject to
significant fines and penalties and may lead to a company being
excluded from participating in federal health care programs.
False Claims Laws. The federal False Claims
Act prohibits knowingly filing a false claim, knowingly causing
the filing of a false claim, or knowingly using false statements
to obtain payment from the federal government. Under the False
Claims Act, such suits are known as “qui tam” actions,
and those who bring such suits. Individuals may file suit on
behalf of the government share in any amounts received by the
government pursuant to a settlement. In addition, certain states
have enacted laws modeled after the federal False Claims Act
under the Deficit Reduction Act of 2005, the federal government
created financial incentives for states to enact false claims
laws consistent with the federal False Claims Act. As more
states enact such laws, we expect the number of qui tam lawsuits
to increase. Qui tam actions have increased significantly in
recent years, causing greater numbers of healthcare companies to
have to defend a false claims action, pay fines or be excluded
from Medicare, Medicaid or other federal or state government
healthcare programs as a result of investigations arising out of
such actions.
HIPAA. Two federal crimes were created under
the Health Insurance Portability and Accountability Act of 1996,
or HIPAA: healthcare fraud and false statements relating to
healthcare matters. The healthcare fraud statute prohibits
knowingly and willfully executing a scheme to defraud any
healthcare benefit program, including private payors. The false
statements statute prohibits knowingly and willfully falsifying,
concealing or covering up a material fact or making any
materially false, fictitious or fraudulent statement in
connection with the delivery of or payment for healthcare
benefits, items or services.
Health
Information Privacy and Security
Individually identifiable health information is subject to an
array of federal and state regulation. Federal rules promulgated
pursuant to HIPAA regulate the use and disclosure of health
information by “covered entities.” Covered entities
include individual and institutional health care providers from
which we may receive individually identifiable health
information. These regulations govern, among other things, the
use and disclosure of health information for research purposes,
and require the covered entity to obtain the written
authorization of the individual before using or disclosing
health information for research. Failure of the covered entity
to obtain such
24
authorization could subject the covered entity to civil and
criminal penalties. We may experience delays and complex
negotiations as we deal with each entity’s differing
interpretation of the regulations and what is required for
compliance. Also, where our customers or contractors are covered
entities, including hospitals, universities, physicians or
clinics, we may be required by the HIPAA regulations to enter
into “business associate” agreements that subject us
to certain privacy and security requirements. In addition, many
states have laws that apply to the use and disclosure of health
information, and these laws could also affect the manner in
which we conduct our research and other aspects of our business.
Such state laws are not preempted by the federal privacy law
where they afford greater privacy protection to the individual.
While activities to assure compliance with health information
privacy laws are a routine business practice, we are unable to
predict the extent to which our resources may be diverted in the
event of an investigation or enforcement action with respect to
such laws.
Foreign
Regulation
Whether or not we obtain FDA approval for a product, we must
obtain approval of a product by the applicable regulatory
authorities of foreign countries before we can commence clinical
trials or marketing of the product in those countries. The
approval process varies from country to country, and the time
may be longer or shorter than that required for FDA approval.
The requirements governing the conduct of clinical trials,
product licensing, pricing, and reimbursement also vary greatly
from country to country. Although governed by the applicable
country, clinical trials conducted outside of the United States
typically are administered under a three-phase sequential
process similar to that discussed above for pharmaceutical
products.
European
Union Regulation
Medical Device Regulation. Our Microcyn
products are classified as medical devices in the European
Union. In order to sell our medical device products within the
European Union, we are required to comply with the requirements
of the Medical Devices Directive, or MDD, and its national
implementations, including affixing CE Marks on our
products. In order to comply with the MDD, we must meet certain
requirements relating to the safety and performance of our
products and, prior to marketing our products, we must
successfully undergo verification of our product’s
regulatory compliance, or conformity assessment.
Medical devices are divided into three regulatory classes:
Class I, Class IIb and Class III. The nature of
the conformity assessment procedures depends on the regulatory
class of the product. We executed the conformity assessment for
production quality assurance for Class IIb products for
Dermacyn Wound Care. Compliance with production quality
assurance is audited every year by a private entity certified by
government regulators. In order to comply with the examination,
we completed, among other things, a risk analysis and presented
clinical data, which demonstrated that our products met the
performance specifications claimed by us, provided sufficient
evidence of adequate assessment of unwanted side effects and
demonstrated that the benefits to the patient outweigh the risks
associated with the device. We will be subject to continued
supervision and will be required to report any serious adverse
incidents to the appropriate authorities. We will also be
required to comply with additional national requirements that
are beyond the scope of the MDD.
We received our CE certificate for Dermacyn Wound Care as a
Class IIb medical device in February 2005. There can be no
assurance that we will be able to maintain the requirements
established for CE Marks for any or all of our products or that
we will be able to produce these products in a timely and
profitable manner while complying with the requirements of the
MDD and other regulatory requirements.
Marketing Authorizations for Drugs. In order
to obtain marketing approval of any of our drug products in
Europe, we must submit for review an application similar to a
U.S. NDA to the relevant authority. In contrast to the
United States, where the FDA is the only authority that
administers and approves NDAs, in Europe there are multiple
authorities that administer and approve these applications.
Marketing authorizations in Europe expire after five years but
may be renewed.
We believe that our Microcyn-based drugs will be reviewed by the
Committee for Medicinal Products for Human Use, or CHMP, on
behalf of the European Medicines Agency, or EMEA. Based upon the
review of the CHMP, the EMEA provides an opinion to the European
Commission on the safety, quality and efficacy of the drug. The
decision to grant or refuse an authorization is made by the
European Commission.
25
Approval of applications can take several months to several
years, or may be denied. This approval process can be affected
by many of the same factors relating to safety, quality and
efficacy as in the approval process for NDAs in the United
States. As in the United States, European drug regulatory
authorities can require us to perform additional non-clinical
studies and clinical trials. The need for such studies or
trials, if imposed, may delay marketing approval and involve
unanticipated costs. Inspection of clinical investigation sites
by a competent authority may also be required as part of the
regulatory approval procedure. In addition, as a condition of
marketing approval, regulatory agencies in Europe may require
post-marketing surveillance to monitor for adverse effects, or
other additional studies as deemed appropriate. The terms of any
approval, including labeling content, may be more restrictive
than expected and could affect the marketability of a product.
In addition, after approval for the initial indication, further
clinical studies are usually necessary to gain approval for any
additional indications.
European GMP. In the European Union, the
manufacture of pharmaceutical products and clinical trial
supplies is subject to good manufacturing practice, or GMP, as
set forth in the relevant laws and guidelines. Compliance with
GMP is generally assessed by the competent regulatory
authorities. They may conduct inspections of relevant
facilities, and review manufacturing procedures, operating
systems and personnel qualifications. In addition to obtaining
approval for each product, in many cases each drug manufacturing
facility must be approved. Further inspections may occur over
the life of the product.
Mexico
The MOH is the authority in charge of sanitary controls in
Mexico. Sanitary controls are a group of practices related to
the orientation, education, testing, verification and
application of security measures and sanctions exercised by the
MOH. The MOH acts by virtue of the Federal Commission for the
Protection against Sanitary Risks, or COFEPRIS, a decentralized
entity of the MOH whose mission is to protect the population
against sanitary risks, by means of centralized sanitary
regulations, controls and by raising public awareness.
The MOH is responsible for the issuance of Official Mexican
Standards and specifications for drugs subject to the provisions
of the General Health Law, which govern the process and
specifications of drugs, including the obtaining, preparation,
manufacturing, maintenance, mixture, conditioning, packaging,
handling, transport, distribution, storage and supply of
products to the public at large. In addition, a medical device
is defined as a device that may contain antiseptics or
germicides used in surgical practice or in the treatment of
continuity solutions, skin injuries or its attachments.
Regulations applicable to medical devices and drugs are divided
into two sections: the business that manufactures the medical
device or drug and the product itself.
Manufacturing a Medical Device or Drug. Under
the General Health Law, a business that manufactures drugs is
either required to obtain a Sanitary Authorization or to file an
Operating Notice. Our Mexico subsidiary is considered a business
that manufactures medical devices and therefore is not subject
to a Sanitary Authorization, but rather only an Operating Notice.
In addition to its Operating Notice, our Mexico subsidiary has
obtained a “Good Processing Practices Certificate”
issued by COFEPRIS, which demonstrates that the manufacturing of
Microcyn at the facility located in Zapopan, Mexico, operates in
accordance with the applicable official standards.
Commercialization of Drugs and Medical
Devices. Drugs and medical devices should be
commercialized in appropriate packaging containing labels
printed in accordance with specific official standards. For
medical devices, there are no specific standards or regulations
related to the labeling of the product, but rather only a
general standard related to the labeling for all types of
products to be commercialized in Mexico. Advertising of medical
devices is regulated in the General Health Law and in the
specific regulations of the General Health Law related to
advertising. Generally, the advertising of medical devices is
subject to a permit only in the case that such advertising is
directed to the general public.
Medical Devices and Drugs as a Product. To
produce, sell or distribute medical devices, a Sanitary Registry
is required in accordance with the General Health Law and the
Regulation for Drugs. Such registry is granted for a term of
five years, and this term may be extended. The Sanitary Registry
may be revoked if the interested party does not request the
extension in the term or the product or the manufacturer or the
raw material is changed without the permission of the MOH.
26
The MOH classifies the medical devices in three classes:
|
|
|
| |
•
|
Class I. Devices for which safety and
effectiveness have been duly proved and are generally not used
inside the body;
|
| |
| |
•
|
Class II. Devices that may vary with
respect to the material used for its fabrication or in its
concentration and generally used in the inside of the body for a
period no greater than 30 days; and
|
| |
| |
•
|
Class III. New devices or recently
approved devices in the medical practice or those used inside
the body and which shall remain inside the body for a period
greater than 30 days.
|
Violation of these regulations may result in the revocation of
the registrations or approvals, and, in addition, economic
fines. In some cases, such violations may constitute criminal
actions.
In addition, regulatory approval of prices is required in most
countries other than the United States, which could result in
lengthy negotiations delaying our ability to commercialize our
products. We face the risk that the prices which result from the
regulatory approval process would be insufficient to generate an
acceptable return.
Employees
As of March 31, 2007, we had 69 full-time
employees, including 15 in manufacturing, 12 in research and
development, 6 in regulatory and clinical, 12 in sales and
marketing and 14 in executive or administrative functions in the
U.S., 3 in administrative functions in Europe, 5 in
administrative functions in Mexico, and 2 in information
technology function. None of our employees is covered by
collective bargaining arrangements, and we consider our
relationship with our employees to be good.
Available
Information
Our website is located at www.oculusis.com. We make available
free of charge on our website our annual reports on
Form 10-K,
quarterly reports on
Form 10-Q,
current reports on
Form 8-K
and amendments to those reports, as soon as reasonably
practicable after we electronically file or furnish such
materials to the Securities and Exchange Commission. Our website
and the information contained therein or connected thereto are
not intended to be incorporated into this Annual Report on
Form 10-K.
Factors
that May Affect Results
Risks
Related to Our Business
We
have a history of losses, we expect to continue to incur losses
and we may never achieve profitability.
We have incurred significant net losses in each fiscal year
since our inception, including losses of $19.8 million,
$21.1 million and $16.5 million for the years ended
March 31, 2007, 2006 and 2005 respectively. Our accumulated
deficit as of March 31, 2007 was $70.5 million. We
have yet to demonstrate that we can generate sufficient sales of
our products to become profitable. The extent of our future
operating losses and the timing of profitability are highly
uncertain, and we may never achieve profitability. Even if we do
generate significant revenues from our product sales, we expect
that increased operating expenses will result in significant
operating losses in the near term as we, among other things:
|
|
|
| |
•
|
conduct preclinical studies and clinical trials on our products
and product candidates;
|
| |
| |
•
|
seek FDA clearance to market Microcyn as a drug in the United
States;
|
| |
| |
•
|
increase our research and development efforts to enhance our
existing products, commercialize new products and develop new
product candidates;
|
| |
| |
•
|
establish additional and expand existing manufacturing
facilities; and
|
| |
| |
•
|
grow our sales and marketing capabilities in the United States
and internationally.
|
27
As a result of these activities, we will need to generate
significant revenue in order to achieve profitability and may
never become profitable. We must also maintain specified cash
reserves in connection with our loan and security agreement
which may limit our investment opportunities. Failure to
maintain these reserves could result in our secured lenders
foreclosing against our assets or imposing significant
restrictions on our operations. Even if we do achieve
profitability, we may not be able to sustain or increase
profitability on an ongoing basis.
Without raising additional capital, we would curtail certain
operational activities in order to reduce costs. We cannot
provide any assurance that we will secure any commitments for
new financing on acceptable terms, if at all.
Because
all of our products are based on our Microcyn platform
technology, we will need to generate sufficient revenues from
the sale of Microcyn to execute our business plan.
All of our products are based on our Microcyn platform
technology, and we do not have any non-Microcyn product
candidates that will generate revenues in the foreseeable
future. Accordingly, we expect to derive substantially all of
our future revenues from sales of our current Microcyn products.
We have only been selling our products since July 2004, and
substantially all of our historical product revenues have been
from sales of Microcyn in Mexico. Although we began selling in
Europe in October 2004, in the United States in June 2005, and
in India in July 2006, our product revenues outside of Mexico
were not significant prior to fiscal year 2007. For example,
product revenues from countries outside of Mexico were just 9%
of our product revenues for the year ended March 31, 2006.
However, during the year ended March 31, 2007, the
percentage of product revenues from outside of Mexico increased
to 32%. Microcyn has not been adopted as a standard of care for
wound treatment in any country and may not gain acceptance among
physicians, nurses, patients, third-party payors and the medical
community. Existing protocols for wound care are well
established within the medical community and tend to vary
geographically, and healthcare providers may be reluctant to
alter their protocols to include the use of Microcyn. If
Microcyn does not achieve an adequate level of acceptance, we
will not generate sufficient revenues to become profitable. We
recently decreased our sales and marketing activities in Europe
and Mexico, which could materially affect our revenues in the
geographic areas in the future.
Our
inability to raise additional capital on acceptable terms in the
future may cause us to curtail certain operational activities,
including regulatory trials, sales and marketing, and
international operations, in order to reduce costs and sustain
the business, and would have a material adverse effect on our
business and financial condition.
We expect capital outlays and operating expenditures to increase
over the next several years as we work to conduct regulatory
trials, commercialize our products and expand our
infrastructure. We have entered into debt financing arrangements
which are secured by all of our assets. We may need to raise
additional capital to, among other things:
|
|
|
| |
•
|
fund our clinical trials and preclinical studies;
|
| |
| |
•
|
sustain commercialization of our current products or new
products;
|
| |
| |
•
|
expand our manufacturing capabilities;
|
| |
| |
•
|
increase our sales and marketing efforts to drive market
adoption and address competitive developments;
|
| |
| |
•
|
acquire or license technologies; and
|
| |
| |
•
|
finance capital expenditures and our general and administrative
expenses.
|
Our present and future funding requirements will depend on many
factors, including:
|
|
|
| |
•
|
the progress and timing of our clinical trials;
|
| |
| |
•
|
the level of research and development investment required to
maintain and improve our technology position;
|
| |
| |
•
|
cost of filing, prosecuting, defending and enforcing patent
claims and other intellectual property rights;
|
| |
| |
•
|
our efforts to acquire or license complementary technologies or
acquire complementary businesses;
|
28
|
|
|
| |
•
|
changes in product development plans needed to address any
difficulties in commercialization;
|
| |
| |
•
|
competing technological and market developments; and
|
| |
| |
•
|
changes in regulatory policies or laws that affect our
operations.
|
If we raise additional funds by issuing equity securities,
dilution to our stockholders could result. Any equity securities
issued also may provide for rights, preferences or privileges
senior to those of holders of our common stock. If we raise
additional funds by issuing debt securities, these debt
securities would have rights, preferences and privileges senior
to those of holders of our common stock, and the terms of the
debt securities issued could impose significant restrictions on
our operations. If we raise additional funds through
collaborations and licensing arrangements, we might be required
to relinquish significant rights to our technologies or
products, or grant licenses on terms that are not favorable to
us. A failure to obtain adequate funds may cause us to curtail
certain operational activities, including regulatory trials,
sales and marketing, and international operations, in order to
reduce costs and sustain the business, and would have a material
adverse effect on our business and financial condition.
We do
not have the necessary regulatory approvals to market Microcyn
as a drug in the United States.
We have obtained three 510(k) clearances in the United States
that permit us to sell Microcyn as a medical device to clean,
moisten and debride wounds. However, we do not have the
necessary regulatory approvals to market Microcyn in the United
States as a drug, which we will need to obtain in order to
execute our business plan. Before we are permitted to sell
Microcyn as a drug in the United States, we must, among other
things, successfully complete additional preclinical studies and
well-controlled clinical trials, submit a New Drug Application,
or NDA, to the FDA and obtain FDA approval. In July 2006, we
completed a controlled clinical trial for pre-operative skin
preparation. After completion of this trial, the FDA advised us
that it is considering adopting new heightened performance
requirements for evaluating efficacy of products designed to be
used in pre-operative skin preparation such as ours. In
discussions with the FDA, the FDA has not provided us with the
definitive timing for, or parameters of, any such requirements,
and has informally stated that it is uncertain during what time
frame it will be able to do so. We plan to continue our
discussions with the FDA regarding the possible timing and
parameters of any new guidelines for evaluating efficacy for
pre-operative skin preparations. Depending on the ultimate
position of the FDA regarding performance criteria for
pre-operative skin preparations, we may reassess our priorities,
clinical timelines and schedules for pursuing a pre-operative
skin preparation indication or may decide not to pursue this
indication. We also intend to seek FDA approval for the use of
Microcyn to treat infections in wounds.
We have sponsored the majority of physicians performing
physician clinical studies of Microcyn and in some cases, the
physicians who performed these studies also hold equity in our
company. The physician clinical studies were performed in the
United States, Mexico and Italy, and used various endpoints,
methods and controls. These studies were not intended to be
rigorously designed or controlled clinical trials and, as such,
did not have all of the controls required for clinical trials
used to support an NDA submission to the FDA in that they did
not include blinding, randomization, predefined clinical
endpoints, use of placebo and active control groups or
U.S. good clinical practice requirements. Consequently, the
results of these physician clinical studies may not be used by
us to support an NDA submission for Microcyn to the FDA. In
addition, any results obtained from clinical trials designed to
support an NDA submission for Microcyn to the FDA may not be as
favorable as results from such physician clinical studies and
otherwise may not be sufficient to support an NDA submission or
FDA approval of any Microcyn NDA.
The FDA approval process is expensive and uncertain, requires
detailed and comprehensive scientific and other data and
generally takes several years. Despite the time and expense
exerted, approval is never guaranteed. We do not know whether we
will obtain favorable results in our preclinical and clinical
studies or whether we will obtain the necessary regulatory
approvals to market Microcyn as a drug in the United States. We
anticipate that obtaining approval for the use of Microcyn to
treat infections in wounds in the United States will take
several years. Even if we obtain FDA approval to sell Microcyn
as a drug, we may not be able to successfully commercialize
Microcyn as a drug in the United States and may never recover
the substantial costs we have invested in the development of our
Microcyn products.
29
Delays
or adverse results in clinical trials could result in increased
costs to us and delay our ability to generate
revenue.
Clinical trials can be long and expensive, and the outcome of
clinical trials is uncertain and subject to delays. It may take
several years to complete clinical trials, if at all, and a
product candidate may fail at any stage of the clinical trial
process. The length of time required varies substantially
according to the type, complexity, novelty and intended use of
the product candidate. Interim results of a preclinical study or
clinical trial do not necessarily predict final results, and
acceptable results in preclinical studies or early clinical
trials may not be repeatable in later subsequent clinical
trials. The commencement or completion of any of our clinical
trials may be delayed or halted for a variety of reasons,
including the following:
|
|
|
| |
•
|
FDA requirements for approval, including requirements for
testing efficacy or safety, may change;
|
| |
| |
•
|
the FDA or other regulatory authorities do not approve a
clinical trial protocol;
|
| |
| |
•
|
patients do not enroll in clinical trials at the rate we expect;
|
| |
| |
•
|
delays in reaching agreement on acceptable clinical trial
agreement terms with prospective sites;
|
| |
| |
•
|
delays in obtaining institutional review board approval to
conduct a study at a prospective site;
|
| |
| |
•
|
third party clinical investigators do not perform our clinical
trials on our anticipated schedule or consistent with the
clinical trial protocol and good clinical practices, or the
third party organizations do not perform data collection and
analysis in a timely or accurate manner;
|
| |
| |
•
|
governmental regulations or administrative actions are
changed; and
|
| |
| |
•
|
insufficient funds to continue our clinical trials.
|
We do not know whether our existing or any future clinical
trials will demonstrate safety and efficacy sufficiently to
result in additional FDA approvals. While a number of physicians
have conducted clinical studies assessing the safety and
efficacy of Microcyn for various indications, the data from
these studies is not sufficient to support approval of Microcyn
as a drug in the United States. In addition, further studies and
trials could show different results. For example, in an
independent physician study of 10 patients in which
procedures were not fully delineated, published in February
2007, four patients discontinued treatment with Demacyn due to
pain, and beneficial change in wound microbiology was found in
only one of the six remaining patients. We will be required to
conduct additional clinical trials prior to seeking approval of
Microcyn for additional indications. Our failure to adequately
demonstrate the safety and efficacy of our product candidates to
the satisfaction of the FDA will prevent our receipt of FDA
approval for additional indications and, ultimately, impact
commercialization of our products in the United States. If we
experience significant delays or adverse results in clinical
trials, our financial results and the commercial prospects for
products based on Microcyn will be harmed, our costs would
increase and our ability to generate revenue would be delayed.
One of
our non-commercialized products, when tested by the
U.S. Environmental Protection Agency, or EPA, did not meet
certain efficacy standards based on an EPA test protocol that
used parameters that differed from those parameters previously
used by us when we originally registered this product as an EPA
registered disinfectant product.
In October 2004, after EPA review of our registration filing,
including the results of disinfectant efficacy testing conducted
by an independent laboratory retained by us, we obtained EPA
authorization, or registration, for the distribution and sale of
our Microcyn-based product, which we call Cidalcyn, as a
hospital grade disinfectant. Although we have not commercialized
Cidalcyn, we previously provided samples to potential marketing
partners and other entities for product evaluation.
Subsequently, in July 2006, we were informed by the EPA that in
more recent tests conducted by the EPA, Cidalcyn did not meet
efficacy standards when tested against three specified pathogens
(Pseudomonas aeruginosa, Staphylococcus aureus and
Mycobacterium tuberculosis) when used according to label
directions. We believe the EPA test protocol utilizes a
bacterial culture to challenge a disinfectant in a test method
which does not replicate a human wound environment and which is
not used to evaluate the safety or efficacy of wound care
products by the FDA or CE Mark. We believe the EPA test made use
of a bacterial culture
30
which contained a significantly higher concentration of
pathogens than the culture used in the independent test, the
results of which we submitted to the EPA for registration
purposes. This increased concentration of bacteria might have
overwhelmed our Cidalcyn product. Subsequent testing we have
conducted appears to have confirmed the EPA’s results
against two of the three pathogens. Based on the EPA’s own
testing, the EPA strongly recommended that we immediately recall
all Cidalcyn distributed on and after September 28, 2005.
Accordingly, we promptly and voluntarily ceased all distribution
of Cidalcyn to end users, and we are not providing the product
to distributors or retailers for re-distribution to third
parties or end users; we have ceased promoting Cidalcyn; and we
have contacted the entities and small number of individuals in
the United States who are not our employees, to whom the
Cidalcyn product had been provided for evaluation purposes
during the one-year period (the product’s shelf-life) prior
to our receipt of the EPA’s recent notification to ensure
they have been informed not to use any remaining quantities they
might have in their possession. In August 2006, we received a
“show cause” letter from the EPA stating that it was
prepared to file a civil administrative complaint against us for
violation of federal pesticide legislation in connection with
the sale or distribution of a pesticide that did not meet the
label’s efficacy claims, and it gave us the opportunity to
advise the EPA of any factors we believe the EPA should consider
before issuing a civil complaint. We have engaged in discussions
with the EPA and since that time and worked cooperatively with
the EPA to resolve this matter. Unless and until we provide new
information to support the original label claims of Cidalcyn to
the EPA, there will not be any sales or other distributions of
the product in the United States as a hospital grade
disinfectant. On April 5, 2007, we entered into a Consent
Agreement and Final Order with the EPA allowing us to amend our
EPA registration to a food sanitizer and pay a $20,800 fine
without admitting or denying any wrongdoing.
If we
fail to obtain, or experience significant delays in obtaining
additional regulatory clearances or approvals to market our
current or future products, we may be unable to commercialize
these products.
Developing, testing, manufacturing, marketing and selling of
medical technology products are subject to extensive regulation
by numerous governmental authorities in the United States and
other countries. The process of obtaining regulatory clearance
and approval of medical technology products is costly and time
consuming. Even though the underlying product formulation may be
the same or similar, our products are subject to different
regulations and approval processes depending upon their intended
use. In the United States, use of Microcyn to cleanse and
debride a wound comes within the medical device regulation
framework, while use of Microcyn to treat infections in wounds
will require us to seek FDA approval of Microcyn as a drug in
the United States.
To obtain regulatory approval of our products as drugs in the
United States, we must first show that our products are safe and
effective for target indications through preclinical studies
(laboratory and animal testing) and clinical trials (human
testing). The FDA generally clears marketing of a medical device
through the 510(k) pre-market clearance process if it is
demonstrated that the new product has the same intended use and
the same or similar technological characteristics as another
legally marketed Class II device, such as a device already
cleared by the FDA through the 510(k) premarket notification
process, and otherwise meets the FDA’s requirements.
Product modifications, including labeling the product for a new
intended use, may require the submission of a new 510(k)
clearance and FDA approval before the modified product can be
marketed.
We do not know whether our products based on Microcyn will
receive approval from the FDA as a drug. The data from clinical
studies of Microcyn conducted by physicians to date will not
satisfy the FDA’s regulatory criteria for approval of an
NDA. In order for us to seek approval for the use of Microcyn as
a drug in the treatment of infections in wounds, we will be
required to conduct additional preclinical and clinical trials
and submit applications for approval to the FDA. For example, we
are currently planning to conduct a pilot study of Microcyn for
the treatment of wound infections, and we will need to conduct
additional non-clinical and well-controlled clinical trials in
order to generate data to support FDA approval of Microcyn for
this indication.
The outcomes of clinical trials are inherently uncertain. In
addition, we do not know whether the necessary approvals or
clearances will be granted or delayed for future products. The
FDA could request additional information or clinical testing
that could adversely affect the time to market and sale of
products as drugs. If we do not obtain the requisite regulatory
clearances and approvals, we will be unable to commercialize our
products as drugs or devices and may never recover any of the
substantial costs we have invested in the development of
Microcyn.
31
Distribution of our products outside the United States is
subject to extensive government regulation. These regulations,
including the requirements for approvals or clearance to market,
the time required for regulatory review and the sanctions
imposed for violations, vary from country to country. We do not
know whether we will obtain regulatory approvals in such
countries or that we will not be required to incur significant
costs in obtaining or maintaining these regulatory approvals. In
addition, the export by us of certain of our products that have
not yet been cleared for domestic commercial distribution may be
subject to FDA export restrictions. Failure to obtain necessary
regulatory approvals, the restriction, suspension or revocation
of existing approvals or any other failure to comply with
regulatory requirements would have a material adverse effect on
our future business, financial condition, and results of
operations.
If our
products do not gain market acceptance, our business will suffer
because we might not be able to fund future
operations.
A number of factors may affect the market acceptance of our
products or any other products we develop or acquire, including,
among others:
|
|
|
| |
•
|
the price of our products relative to other treatments for the
same or similar treatments;
|
| |
| |
•
|
the perception by patients, physicians and other members of the
health care community of the effectiveness and safety of our
products for their indicated applications and treatments;
|
| |
| |
•
|
our ability to fund our sales and marketing efforts; and
|
| |
| |
•
|
the effectiveness of our sales and marketing efforts.
|
If our products do not gain market acceptance, we may not be
able to fund future operations, including developing, testing
and obtaining regulatory approval for new product candidates and
expanding our sales and marketing efforts for our approved
products, which would cause our business to suffer.
We
have agreed to change the brand name of our product in Mexico,
which may result in the loss of any brand recognition that we
have established with users of our products.
In accordance with the settlement of a trademark infringement
lawsuit filed against us in Mexico, we have agreed to stop using
the name Microcyn60 in Mexico by September 2007. In addition, in
May 2006, a complaint was filed against us for trademark
confusion in connection with the same tradename, and we are in
settlement negotiations concerning such claim. We have marketed
our products in Mexico under the brand name of Microcyn60 since
2004. During the year ended March 31, 2007, 68% of our
product revenues were derived from Mexico. As a result of our
agreement to change our product name, we may lose the benefit of
the brand name recognition we have generated in the region and
our product sales in Mexico could decline. In locations where we
have distributed our products, we believe that the brand names
of those products have developed name recognition among
consumers who purchase them. Any change to the brand name of our
other products may cause us to lose such name recognition, which
may lead to confusion in the marketplace and a decline in sales
of our products.
If our
competitors develop products similar to Microcyn, we may need to
modify or alter our business strategy, which may delay the
achievement of our goals.
Competitors may develop products with similar characteristics as
Microcyn. Such similar products marketed by larger competitors
can hinder our efforts to penetrate the market. As a result, we
may be forced to modify or alter our business and regulatory
strategy and sales and marketing plans, as a response to changes
in the market, competition and technology limitations, among
others. Such modifications may pose additional delays in
achieving our goals.
32
We
intend to license or collaborate with third parties in various
potential markets, and events involving these strategic partners
or any future collaborations could delay or prevent us from
developing or commercializing products.
Our business strategy and our short- and long-term operating
results will depend in part on our ability to execute on
existing strategic collaborations and to license or partner with
new strategic partners. We believe collaborations allow us to
leverage our resources and technologies and to access markets
that are compatible with our own core areas of expertise while
avoiding the cost of establishing a direct sales force in each
market. We may incur significant costs in the use of third
parties to identify and assist in establishing relationships
with potential collaborators.
To penetrate our target markets, we may need to enter into
additional collaborative agreements to assist in the development
and commercialization of future products. For example, depending
upon our analysis of the time and expense involved in obtaining
FDA approval to sell a product to treat open wounds, we may
choose to license our technology to a third party as opposed to
pursuing commercialization ourselves. Establishing strategic
collaborations is difficult and time-consuming. Potential
collaborators may reject collaborations based upon their
assessment of our financial, regulatory or intellectual property
position and our internal capabilities. Our discussions with
potential collaborators may not lead to the establishment of new
collaborations on favorable terms. We have limited control over
the amount and timing of resources that our current
collaborators or any future collaborators devote to our
collaborations or potential products. These collaborators may
breach or terminate their agreements with us or otherwise fail
to conduct their collaborative activities successfully and in a
timely manner. Further, our collaborators may not develop or
commercialize products that arise out of our collaborative
arrangements or devote sufficient resources to the development,
manufacture, marketing or sale of these products. By entering
into a collaboration, we may preclude opportunities to
collaborate with other third parties who do not wish to
associate with our existing third party strategic partners.
Moreover, in the event of termination of a collaboration
agreement, termination negotiations may result in less favorable
terms.
If we
are unable to expand our direct domestic sales force, we may not
be able to successfully sell our products in the United
States.
We have very limited commercialization capability and make
Microcyn-based products available primarily through our website,
one national distributor and several regional distributors. We
plan for a more aggressive commercialization and product launch
in the event we obtain drug approval from the FDA. Developing a
sales force is expensive and time consuming, and the lack of
qualified sales personnel could delay or limit the success of
our product launch. Our domestic sales force, if established,
will be competing with the sales operations of our competitors,
which are better funded and more experienced. We may not be able
to develop domestic sales capacity on a timely basis or at all.
We may
incur significant liabilities in connection with our
relationship with a former distributor in Mexico.
On June 16, 2005, we entered into a series of agreements
with Quimica Pasteur, or QP, a Mexico-based distributor of
pharmaceutical products to hospitals and health care entities
owned or operated by the Mexican Ministry of Health, or MOH.
These agreements provided, among other things, for QP to act as
our exclusive distributor of Microcyn to the MOH for a period of
three years. We were granted an option to acquire all except a
minority share of the equity of QP directly from its principals.
In addition, two of our employees were appointed as officers of
QP, which resulted in the establishment of financial control of
QP by our company under applicable accounting literature. As a
result of our agreements, we were required to consolidate
QP’s operations with our financial results for a portion of
our year ended March 31, 2006. In connection with our audit
of QP’s financial statements in late 2005, we were made
aware of a number of facts that suggested that QP or its
principals may have engaged in some form of tax avoidance
practice in Mexico prior to the execution of the agreements
between our company and QP. We did not discover these facts
prior to our execution of these agreements or for several months
thereafter. Our prior independent auditors informed us that we
did not have effective anti-fraud programs designed to detect
the type of activities in which QP’s principals engaged or
the personnel to effectively evaluate and determine the
appropriate accounting for non-routine or complex accounting
transactions. Our audit committee engaged an outside law firm to
conduct an investigation whose findings implicated QP’s
principals in a systemic tax
33
avoidance practice prior to June 16, 2005. We estimate that
QP’s liability for taxes, interest and penalties related to
these practices could amount to $7 million or more. Based
on the results of this investigation, we terminated our
agreements with QP effective March 26, 2006.
Although we do not believe that we are responsible for any tax
avoidance practices of QP’s principals prior to
June 16, 2005, the Mexican taxing authority could make a
claim against us or our Mexican subsidiary. We have been
informed by counsel in Mexico that the statute of limitations,
including for actions for fraud, is five years from
March 31, 2006.
Our
dependence on distributors for sales could limit or prevent us
from selling our products and from realizing long-term revenue
growth.
We currently depend on distributors to sell Microcyn in the
United States, Europe and other countries and intend to continue
to sell our products primarily through distributors in Europe
and the United States for the foreseeable future. If we are
unable to expand our direct sales force, we will continue to
rely on distributors to sell Microcyn. Our existing distribution
agreements are generally short-term in duration, and we may need
to pursue alternate distributors if the other parties to these
agreements terminate or elect not to renew their agreements. If
we are unable to retain our current distributors for any reason,
we must replace them with alternate distributors experienced in
supplying the wound care market, which could be time-consuming
and divert management’s attention from other operational
matters. In addition, we will need to attract additional
distributors to expand the geographic areas in which we sell
Microcyn. Distributors may not commit the necessary resources to
market and sell our products to the level of our expectations,
which could harm our ability to generate revenues. In addition,
some of our distributors may also sell products that compete
with ours. In some countries, regulatory licenses must be held
by residents of the country. For example, the regulatory
approval for one product in India is owned and held by our
Indian distributor. If the licenses are not in our name or under
our control, we might not have the power to ensure their ongoing
effectiveness and use by us. If current or future distributors
do not perform adequately, or we are unable to locate
distributors in particular geographic areas, we may not realize
long-term revenue growth.
We
depend on a contract sales force to sell our products in
Mexico.
We currently depend on a contract sales force to sell Microcyn
in Mexico. Our existing agreement is short-term in duration and
can be terminated by either party upon 30 days written
notice. If we are unable to retain our current agreement for any
reason, we may need to build our own internal sales force or
find an alternate source for contract sales people. We may be
unable to find an alternate source, or the alternate
source’s sales force may not generate sufficient revenue.
If our current or future contract sales force does not perform
adequately, we may not realize long-term revenue growth in
Mexico.
If we
fail to comply with ongoing regulatory requirements, or if we
experience unanticipated problems with our products, these
products could be subject to restrictions or withdrawal from the
market.
Regulatory approvals or clearances that we currently have and
that we may receive in the future are subject to limitations on
the indicated uses for which the products may be marketed, and
any future approvals could contain requirements for potentially
costly post-marketing
follow-up
studies. If the FDA determines that our promotional materials or
activities constitute promotion of an unapproved use or we
otherwise fail to comply with FDA regulations, we may be subject
to regulatory enforcement actions, including a warning letter,
injunction, seizure, civil fine or criminal penalties. In
addition, the manufacturing, labeling, packaging, adverse event
reporting, storage, advertising, promotion, distribution and
record-keeping for approved products are subject to extensive
regulation. Our manufacturing facilities, processes and
specifications are subject to periodic inspection by the FDA,
European and other regulatory authorities and from time to time,
we may receive notices of deficiencies from these agencies as a
result of such inspections. Our failure to continue to meet
regulatory standards or to remedy any deficiencies could result
in restrictions being imposed on products or manufacturing
processes, fines, suspension or loss of regulatory approvals or
clearances, product recalls, termination of distribution or
product seizures or the need to invest substantial resources to
comply with various existing and new requirements. In the more
egregious cases, criminal sanctions, civil penalties,
disgorgement of profits or closure of our manufacturing
facilities are possible. The subsequent discovery of previously
unknown problems with Microcyn, including adverse events of
34
unanticipated severity or frequency, may result in restrictions
on the marketing of our products, and could include voluntary or
mandatory recall or withdrawal of products from the market.
New government regulations may be enacted and changes in FDA
policies and regulations, their interpretation and enforcement,
could prevent or delay regulatory approval of our products. We
cannot predict the likelihood, nature or extent of adverse
government regulation that may arise from future legislation or
administrative action, either in the United States or abroad.
Therefore, we do not know whether we will be able to continue to
comply with any regulations or that the costs of such compliance
will not have a material adverse effect on our future business,
financial condition, and results of operations. If we are not
able to maintain regulatory compliance, we will not be permitted
to market our products and our business would suffer.
We may
experience difficulties in manufacturing Microcyn, which could
prevent us from commercializing one or more of our
products.
The machines used to manufacture our Microcyn-based products are
complex, use complicated software and must be monitored by
highly trained engineers. Slight deviations anywhere in our
manufacturing process, including quality control, labeling and
packaging, could lead to a failure to meet the specifications
required by the FDA, the EPA, European notified bodies, Mexican
regulatory agencies and other foreign regulatory bodies, which
may result in lot failures or product recalls. In August 2006,
we received a “show cause” letter from the EPA, which
stated that, in tests conducted by the EPA, Cidalcyn was found
to be ineffective in killing specified pathogens when used
according to label directions. We gathered records for review to
determine if there might have been any problems in production of
the lot tested by the EPA. We have also quarantined all
remaining quantities of the production lot in question. If we
are unable to obtain quality internal and external components,
mechanical and electrical parts, if our software contains
defects or is corrupted, or if we are unable to attract and
retain qualified technicians to manufacture our products, our
manufacturing output of Microcyn, or any other product candidate
based on our platform that we may develop, could fail to meet
required standards, our regulatory approvals could be delayed,
denied or revoked, and commercialization of one or more of our
Microcyn-based products may be delayed or foregone.
Manufacturing processes that are used to produce the smaller
quantities of Microcyn needed for our clinical test and current
commercial sales may not be successfully scaled up to allow
production of significant commercial quantities. Any failure to
manufacture our products to required standards on a commercial
scale could result in reduced revenues, delays in generating
revenue and increased costs.
Our
competitive position depends on our ability to protect our
intellectual property and our proprietary
technologies.
Our ability to compete and to achieve and maintain profitability
depends on our ability to protect our intellectual property and
proprietary technologies. We currently rely on a combination of
patents, patent applications, trademarks, trade secret laws,
confidentiality agreements, license agreements and invention
assignment agreements to protect our intellectual property
rights. We also rely upon unpatented know-how and continuing
technological innovation to develop and maintain our competitive
position. These measures may not be adequate to safeguard our
Microcyn technology. In addition, we granted a security interest
in our assets, including our intellectual property, under two
loan and security agreements. If we do not protect our rights
adequately, third parties could use our technology, and our
ability to compete in the market would be reduced.
Although we have filed U.S. and foreign patent applications
related to our Microcyn based products, the manufacturing
technology for making the products, and their uses, only one
patent has been issued from these applications to date.
Our pending patent applications and any patent applications we
may file in the future may not result in issued patents, and we
do not know whether any of our in-licensed patents or any
additional patents that might ultimately be issued by the
U.S. Patent and Trademark Office or foreign regulatory body
will protect our Microcyn technology. Any claims that issue may
not be sufficiently broad to prevent third parties from
producing competing substitutes and may be infringed, designed
around, or invalidated by third parties. Even issued patents may
later be found to be invalid, or may be modified or revoked in
proceedings instituted by third parties before various patent
offices or in courts.
35
The degree of future protection for our proprietary rights is
more uncertain in part because legal means afford only limited
protection and may not adequately protect our rights, and we
will not be able to ensure that:
|
|
|
| |
•
|
we were the first to invent the inventions described in patent
applications;
|
| |
| |
•
|
we were the first to file patent applications for inventions;
|
| |
| |
•
|
others will not independently develop similar or alternative
technologies or duplicate our products without infringing our
intellectual property rights;
|
| |
| |
•
|
any patents licensed or issued to us will provide us with any
competitive advantages;
|
| |
| |
•
|
we will develop proprietary technologies that are
patentable; or
|
| |
| |
•
|
the patents of others will not have an adverse effect on our
ability to do business.
|
The policies we use to protect our trade secrets may not be
effective in preventing misappropriation of our trade secrets by
others. In addition, confidentiality and invention assignment
agreements executed by our employees, consultants and advisors
may not be enforceable or may not provide meaningful protection
for our trade secrets or other proprietary information in the
event of unauthorized use or disclosures. We cannot be certain
that the steps we have taken will prevent the misappropriation
and use of our intellectual property, particularly in foreign
countries where the laws may not protect our proprietary rights
as fully as in the United States. For example, one of our former
contract partners, Nofil Corporation, whom we relied upon to
manufacture our proprietary machines had access to our
proprietary information and we believe undertook the development
and manufacture of the machines to be sold to third parties in
violation of our agreement with such company. We have brought a
claim against Nofil Corporation in the U.S. District Court
for the Northern District of California. We believe that a
former officer of our Mexico subsidiary collaborated in these
acts, misappropriated our trade secrets, and is currently
selling products in Mexico that are competitive with our
products. In addition, we believe that, through the licensor of
the patents that we in-license and who has also assigned patents
to us, a company in Japan obtained one of our patent
applications, translated it into Hangul and filed it under such
company’s and the licensor’s name in South Korea.
These and any other leak of confidential data into the public
domain or to third parties could allow our competitors to learn
our trade secrets.
We are
in a dispute with the Japanese entity that licenses to us
certain rights under Japanese patents, which could result in our
losing such rights and may have a material adverse impact on our
business opportunities in Japan.
In March 2003, we obtained an exclusive license to six issued
Japanese patents and five Japanese published pending patent
applications owned by Coherent Technologies. The issued Japanese
patents and pending Japanese patent applications relate to an
earlier generation of super-oxidized water product with an
acidic pH and not the current commercialized Microcyn. The
patents that cover the method and apparatus for the production
of the earlier generation of super-oxidized water will expire
between 2011 and 2014. In June 2006, we received written notice
from Coherent Technologies advising us that the patent license
was terminated, citing various reasons with which we disagree.
Since that time we have engaged in discussions with Coherent
Technologies concerning the license agreement and our continued
business relationship. Although we do not believe Coherent
Technologies has grounds to terminate the license, we may have
to take legal action to preserve our rights under the license
and to enjoin Coherent Technologies from breaching its terms. We
do not know whether we would prevail in any such action, which
would be costly and time consuming, and we could lose our rights
under the license, which could have a material adverse impact on
our business opportunities in Japan. In addition, we could have
to defend ourselves against infringement claims from Coherent
Technologies in Japan based on their position on termination of
the license.
36
We may
face intellectual property infringement claims that could be
time-consuming, costly to defend and could result in our loss of
significant rights and, in the case of patent infringement
claims, the assessment of treble damages.
On occasion, we may receive notices of claims of infringement,
misappropriation or misuse of other parties’ proprietary
rights. We may have disputes regarding intellectual property
rights with the parties that have licensed those rights to us.
Some claims received from third parties may lead to litigation.
We cannot assure you that we will prevail in these actions, or
that other actions alleging misappropriation or misuse by us of
third-party trade secrets, infringement by us of third-party
patents and trademarks or the validity of our patents, will not
be asserted or prosecuted against us. We may also initiate
claims to defend our intellectual property. For example, we
brought a claim against Nofil Corporation for misappropriation
of our trade secrets and Nofil Corporation filed a
cross-complaint against us in February 2007 claiming ownership
of our technology. Intellectual property litigation, regardless
of outcome, is expensive and time-consuming, could divert
management’s attention from our business and have a
material negative effect on our business, operating results or
financial condition. In addition, the outcome of such litigation
may be unpredictable. If there is a successful claim of
infringement against us, we may be required to pay substantial
damages (including treble damages if we were to be found to have
willfully infringed a third party’s patent) to the party
claiming infringement, develop non-infringing technology, stop
selling our products or using technology that contains the
allegedly infringing intellectual property or enter into royalty
or license agreements that may not be available on acceptable or
commercially practical terms, if at all. Our failure to develop
non-infringing technologies or license the proprietary rights on
a timely basis could harm our business. In addition, modifying
our products to include the non-infringing technologies could
require us to seek re-approval or clearance from various
regulatory bodies for our products, which would be costly and
time consuming. Also, we may be unaware of pending patent
applications that relate to our technology. Parties making
infringement claims on future issued patents may be able to
obtain an injunction that would prevent us from selling our
products or using technology that contains the allegedly
infringing intellectual property, which could harm our business.
In September 2005, a complaint was filed against us in Mexico
claiming trademark infringement with respect to our Microcyn60
mark. To settle this claim we have agreed to cease marketing our
product in Mexico under the name Microcyn60 by September 2007. A
second unrelated claim was filed against us in Mexico in May
2006, claiming trademark infringement with respect to our
Microcyn60 mark in Mexico. We are in discussions with the
claimant to settle the matter.
In addition to the infringement claims in Mexico, we are
currently involved in several pending trademark opposition
proceedings in connection with our applications to register the
marks Microcyn, Oculus Microcyn and Dermacyn in
the European Union, Argentina, Guatemala, Honduras, Nicaragua
and Paraguay. If we are unable to settle these disputes or
prevail in these opposition proceedings, we will not be able to
obtain registrations for the Microcyn, Oculus Microcyn
and Dermacyn marks in those countries, and that may
impair our ability to enforce our trademark rights against
infringers in those countries. Although no such legal
proceedings have been brought or threats of such legal
proceedings have been made, we cannot rule out the possibility
that any of these opposing parties will also file a trademark
infringement lawsuit seeking to prevent our use and seek
monetary damages based on our use of the Microcyn,
Oculus Microcyn and Dermacyn marks in the European
Union, Argentina, Guatemala, Honduras, Nicaragua and Paraguay.
We have also entered into agreements with third parties to
settle trademark opposition proceedings in which we have agreed
to certain restrictions on our use and registration of certain
marks. In March 2006, we entered into an agreement with an
opposing party that places restrictions on the manner in which
we can use and register our Microcyn and Microcyn60
marks in countries where the opposing party has superior
rights, including in Europe and Singapore. These restrictions
include always using Microcyn along with the word
“technology” and another distinctive trademark such as
Cidalcyn, Dermacyn and Vetericyn. In
addition, we have entered into an agreement with an opposing
party in which we agreed to limit our use and registration of
the Microcyn mark in Uruguay to disinfectant, antiseptic
and sterilizing agents. Moreover, we have entered into an
agreement with an opposing party in Europe in which we agreed to
specifically exclude ophthalmologic products for our Oculus
Microcyn application in the European Union.
37
Our
ability to generate revenue will be diminished if we are unable
to obtain acceptable prices or an adequate level of
reimbursement from third-party payors of healthcare
costs.
The continuing efforts of governmental and other third-party
payors, including managed care organizations such as health
maintenance organizations, or HMOs, to contain or reduce costs
of health care may affect our future revenue and profitability,
and the future revenue and profitability of our potential
customers, suppliers and collaborative or license partners and
the availability of capital. For example, in certain foreign
markets, pricing or profitability of prescription
pharmaceuticals is subject to government control. In the United
States, governmental and private payors have limited the growth
of health care costs through price regulation or controls,
competitive pricing programs and drug rebate programs. Our
ability to commercialize our products successfully will depend
in part on the extent to which appropriate coverage and
reimbursement levels for the cost of our Microcyn products and
related treatment are obtained from governmental authorities,
private health insurers and other organizations, such as HMOs.
There is significant uncertainty concerning third-party coverage
and reimbursement of newly approved medical products and drugs.
Third-party payors are increasingly challenging the prices
charged for medical products and services. Also, the trend
toward managed healthcare in the United States and the
concurrent growth of organizations such as HMOs, as well as
legislative proposals to reform healthcare or reduce government
insurance programs, may result in lower prices for or rejection
of our products. The cost containment measures that health care
payors and providers are instituting and the effect of any
health care reform could materially and adversely affect our
ability to generate revenues.
In addition, given ongoing federal and state government
initiatives directed at lowering the total cost of health care,
the United States Congress and state legislatures will likely
continue to focus on health care reform, the cost of
prescription pharmaceuticals and the reform of the Medicare and
Medicaid payment systems. While we cannot predict whether any
proposed cost-containment measures will be adopted, the
announcement or adoption of these proposals could reduce the
price that we receive for our Microcyn products in the future.
We
could be required to indemnify third parties for alleged
infringement, which could cause us to incur significant
costs.
Some of our distribution agreements contain commitments to
indemnify our distributors against liability arising from
infringement of third party intellectual property such as
patents. We may be required to indemnify our customers for
claims made against them or license fees they are required to
pay. If we are forced to indemnify for claims or to pay license
fees, our business and financial condition could be
substantially harmed.
A
significant part of our business is conducted outside of the
United States, exposing us to additional risks that may not
exist in the United States, which in turn could cause our
business and operating results to suffer.
We have international operations in Mexico and Europe. For the
fiscal years ended March 31, 2007, 2006 and 2005,
approximately 78%, 72% and 35%, respectively, of our total
revenue was generated from sales outside of the United States.
Our business is highly regulated for the use, marketing and
manufacturing of our Microcyn products both domestically and
internationally. Our international operations are subject to
risks, including:
|
|
|
| |
•
|
local political or economic instability;
|
| |
| |
•
|
changes in governmental regulation;
|
| |
| |
•
|
changes in import/export duties;
|
| |
| |
•
|
trade restrictions;
|
| |
| |
•
|
lack of experience in foreign markets;
|
| |
| |
•
|
difficulties and costs of staffing and managing operations in
certain foreign countries;
|
| |
| |
•
|
work stoppages or other changes in labor conditions;
|
38
|
|
|
| |
•
|
difficulties in collecting accounts receivables on a timely
basis or at all; and
|
| |
| |
•
|
adverse tax consequences or overlapping tax structures.
|
We plan to continue to market and sale our products
internationally to respond to customer requirements and market
opportunities. We currently have international manufacturing
facilities in Mexico and The Netherlands. Establishing
operations in any foreign country or region presents risks such
as those described above as well as risks specific to the
particular country or region. In addition, until a payment
history is established over time with customers in a new
geography or region, the likelihood of collecting receivables
generated by such operations could be less than our
expectations. As a result, there is a greater risk that reserves
set with respect to the collection of such receivables may be
inadequate. If our operations in any foreign country are
unsuccessful, we could incur significant losses and we may not
achieve profitability.
In addition, changes in policies or laws of the United States or
foreign governments resulting in, among other things, changes in
regulations and the approval process, higher taxation, currency
conversion limitations, restrictions on fund transfers or the
expropriation of private enterprises, could reduce the
anticipated benefits of our international expansion. If we fail
to realize the anticipated revenue growth of our future
international operations, our business and operating results
could suffer.
Our
sales in international markets subject us to foreign currency
exchange and other risks and costs which could harm our
business.
A substantial portion of our revenues are derived from outside
the United States, primarily from Mexico. We anticipate that
revenues from international customers will continue to represent
a substantial portion of our revenues for the foreseeable
future. Because we generate revenues in foreign currencies, we
are subject to the effects of exchange rate fluctuations. The
functional currency of our Mexican subsidiary is the Mexican
Peso, and the functional currency of our subsidiary in The
Netherlands is the Euro. For the preparation of our consolidated
financial statements, the financial results of our foreign
subsidiaries are translated into U.S. dollars on average
exchange rates during the applicable period. If the
U.S. dollar appreciates against the Mexican Peso or the
Euro, as applicable, the revenues we recognize from sales by our
subsidiaries will be adversely impacted. Foreign exchange gains
or losses as a result of exchange rate fluctuations in any given
period could harm our operating results and negatively impact
our revenues. Additionally, if the effective price of our
products were to increase as a result of fluctuations in foreign
currency exchange rates, demand for our products could decline
and adversely affect our results of operations and financial
condition.
The
loss of key members of our senior management team, one of our
directors or our inability to retain highly skilled scientists,
technicians and salespeople could adversely affect our
business.
Our success depends largely on the skills, experience and
performance of key members of our executive management team,
including Hojabr Alimi, our Chief Executive Officer, and a
member of our Board of Directors and Robert Northey, our Vice
President of Research and Development.. The efforts of these
people will be critical to us as we continue to develop our
products and attempt to commercialize products in the chronic
and acute wound care market. If we were to lose one or more of
these individuals, we may experience difficulties in competing
effectively, developing our technologies and implementing our
business strategies.
Our research and development programs depend on our ability to
attract and retain highly skilled scientists and technicians. We
may not be able to attract or retain qualified scientists and
technicians in the future due to the intense competition for
qualified personnel among medical technology businesses,
particularly in the San Francisco Bay Area. We also face
competition from universities and public and private research
institutions in recruiting and retaining highly qualified
personnel. In addition, our success depends on our ability to
attract and retain salespeople with extensive experience in
wound care and close relationships with the medical community,
including physicians and other medical staff. We may have
difficulties locating, recruiting or retaining qualified
salespeople, which could cause a delay or decline in the rate of
adoption of our products. If we are not able to attract and
retain the necessary personnel to accomplish our business
objectives, we may experience constraints that will adversely
affect our ability to support our research, development and
sales programs.
39
We maintain key-person life insurance only on Mr. Alimi. We
may discontinue this insurance in the future, it may not
continue to be available on commercially reasonable terms or, if
continued, it may prove inadequate to compensate us for the loss
of Mr. Alimi’s services.
We may
be unable to manage our future growth effectively, which would
make it difficult to execute our business
strategy.
We may experience periods of rapid growth as we expand our
business, which will likely place a significant strain on our
limited personnel and other resources. Any failure by us to
manage our growth effectively could have an adverse effect on
our ability to achieve our commercialization goals.
Furthermore, we conduct business in a number of geographic
regions and are seeking to expand to other regions. We have not
established a physical presence in many of the international
regions in which we conduct or plan to conduct business, but
rather we manage our business from our headquarters in Northern
California. As a result, we conduct business at all times of the
day and night with limited personnel. If we fail to
appropriately target and increase our presence in these
geographic regions, we may not be able to effectively market and
sell our Microcyn products in these locations or we may not meet
our customers’ needs in a timely manner, which could
negatively affect our operating results.
Future growth will also impose significant added
responsibilities on management, including the need to identify,
recruit, train and integrate additional employees. In addition,
rapid and significant growth will place strain on our
administrative and operational infrastructure, including sales
and marketing and clinical and regulatory personnel. Our ability
to manage our operations and growth will require us to continue
to improve our operational, financial and management controls,
reporting systems and procedures. If we are unable to manage our
growth effectively, it may be difficult for us to execute our
business strategy.
The
wound care industry is highly competitive and subject to rapid
technological change. If our competitors are better able to
develop and market products that are less expensive or more
effective than any products that we may develop, our commercial
opportunity will be reduced or eliminated.
The wound care industry is highly competitive and subject to
rapid technological change. Our success depends, in part, upon
our ability to stay at the forefront of technological change and
maintain a competitive position.
We compete with large healthcare, pharmaceutical and
biotechnology companies, along with smaller or early-stage
companies that have collaborative arrangements with larger
pharmaceutical companies, academic institutions, government
agencies and other public and private research organizations.
Many of our competitors have significantly greater financial
resources and expertise in research and development,
manufacturing, pre-clinical testing, conducting clinical trials,
obtaining regulatory approvals and marketing approved products
than we do. Our competitors may:
|
|
|
| |
•
|
develop and patent processes or products earlier than we will;
|
| |
| |
•
|
develop and commercialize products that are less expensive or
more efficient than any products that we may develop;
|
| |
| |
•
|
obtain regulatory approvals for competing products more rapidly
than we will; and
|
| |
| |
•
|
improve upon existing technological approaches or develop new or
different approaches that render our technology or products
obsolete or non-competitive.
|
As a result, we may not be able to successfully commercialize
any future products.
40
The
success of our research and development efforts may depend on
our ability to find suitable collaborators to fully exploit our
capabilities. If we are unable to establish collaborations or if
these future collaborations are unsuccessful, our research and
development efforts may be unsuccessful, which could adversely
affect our results of operations and financial
condition.
An important element of our business strategy will be to enter
into collaborative or license arrangements under which we
license our Microcyn technology to other parties for development
and commercialization. We expect that while we may initially
seek to conduct initial clinical trials on our drug candidates,
we may need to seek collaborators for a number of our potential
products because of the expense, effort and expertise required
to conduct additional clinical trials and further develop those
potential products candidates. Because collaboration
arrangements are complex to negotiate, we may not be successful
in our attempts to establish these arrangements. If we need
third party assistance in identifying and negotiating one or
more acceptable arrangements, it might be costly. Also, we may
not have products that are desirable to other parties, or we may
be unwilling to license a potential product because the party
interested in it is a competitor. The terms of any arrangements
that we establish may not be favorable to us. Alternatively,
potential collaborators may decide against entering into an
agreement with us because of our financial, regulatory or
intellectual property position or for scientific, commercial or
other reasons. If we are not able to establish collaborative
agreements, we may not be able to develop and commercialize new
products, which would adversely affect our business and our
revenues.
In order for any of these collaboration or license arrangements
to be successful, we must first identify potential collaborators
or licensees whose capabilities complement and integrate well
with ours. We may rely on these arrangements for, not only
financial resources, but also for expertise or economies of
scale that we expect to need in the future relating to clinical
trials, manufacturing, sales and marketing, and for licenses to
technology rights. However, it is likely that we will not be
able to control the amount and timing of resources that our
collaborators or licensees devote to our programs or potential
products. If our collaborators or licensees prove difficult to
work with, are less skilled than we originally expected, or do
not devote adequate resources to the program, the relationship
will not be successful. If a business combination, involving a
collaborator or licensee and a third party were to occur, the
effect could be to diminish, terminate or cause delays in
development of a potential product.
We may
acquire other businesses or form joint ventures that could harm
our operating results, dilute your ownership of us, increase our
debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of
complementary businesses and assets, as well as technology
licensing arrangements. We also intend to pursue strategic
alliances that leverage our core technology and industry
experience to expand our product offerings or distribution. We
have no experience with respect to acquiring other companies and
limited experience with respect to the formation of
collaborations, strategic alliances and joint ventures. If we
make any acquisitions, we may not be able to integrate these
acquisitions successfully into our existing business, and we
could assume unknown or contingent liabilities. Any future
acquisitions by us also could result in significant write-offs
or the incurrence of debt and contingent liabilities, any of
which could harm our operating results. Integration of an
acquired company also may require management resources that
otherwise would be available for ongoing development of our
existing business. We may not identify or complete these
transactions in a timely manner, on a cost-effective basis, or
at all, and we may not realize the anticipated benefits of any
acquisition, technology license, strategic alliance or joint
venture.
To finance any acquisitions, we may choose to issue shares of
our common stock as consideration, which would dilute your
ownership interest in us. If the price of our common stock is
low or volatile, we may not be able to acquire other companies
for stock. Alternatively, it may be necessary for us to raise
additional funds for acquisitions through public or private
financings. Additional funds may not be available on terms that
are favorable to us, or at all.
If we are unable to comply with broad and complex federal and
state fraud and abuse laws, including state and federal
anti-kickback laws, we could face substantial penalties and our
products could be excluded from government healthcare programs.
We are subject to various federal and state laws pertaining to
healthcare fraud and abuse, which include, among other things,
“anti-kickback” laws that prohibit payments to induce
the referral of products and services, and “false
claims” statutes that prohibit the fraudulent billing of
federal healthcare programs. Our operations are subject to the
41
federal anti-kickback statute, a criminal statute that, subject
to certain statutory exceptions, prohibits any person from
knowingly and willfully offering, paying, soliciting or
receiving remuneration, directly or indirectly, to induce or
reward a person either (i) for referring an individual for
the furnishing of items or services for which payment may be
made in whole or in part by a government healthcare program such
as Medicare or Medicaid, or (ii) for purchasing, leasing,
or ordering or arranging for or recommending the purchasing,
leasing or ordering of an item or service for which payment may
be made under a government healthcare program. Because of the
breadth of the federal anti-kickback statute, the Office of
Inspector General of the U.S. Department of Health and
Human Services, or the OIG, was authorized to adopt regulations
setting forth additional exceptions to the prohibitions of the
statute commonly known as “safe harbors.” If all of
the elements of an applicable safe harbor are fully satisfied,
an arrangement will not be subject to prosecution under the
federal anti-kickback statute.
We previously had agreements to pay compensation to our advisory
board members and physicians who conduct clinical trials or
provide other services for us. Currently, these agreements have
been terminated. The agreements may be subject to challenge to
the extent they do not fall within relevant safe harbors under
federal and similar state anti-kickback laws. If our past or
present operations, including, but not limited to, our
consulting arrangements with our advisory board members or
physicians conducting clinical trials on our behalf, or our
promotional or discount programs, are found to be in violation
of these laws, we or our officers may be subject to civil or
criminal penalties, including large monetary penalties, damages,
fines, imprisonment and exclusion from government healthcare
program participation, including Medicare and Medicaid.
In addition, if there is a change in law, regulation or
administrative or judicial interpretations of these laws, we may
have to change our business practices or our existing business
practices could be challenged as unlawful, which could have a
negative effect on our business, financial condition and results
of operations.
Healthcare fraud and abuse laws are complex and even minor,
inadvertent irregularities can potentially give rise to claims
that a statute or regulation has been violated.
The frequency of suits to enforce these laws have increased
significantly in recent years and have increased the risk that a
healthcare company will have to defend a false claim action, pay
fines or be excluded from the Medicare, Medicaid or other
federal and state healthcare programs as a result of an
investigation arising out of such action. We cannot assure you
that we will not become subject to such litigation. Any
violations of these laws, or any action against us for violation
of these laws, even if we successfully defend against it, could
harm our reputation, be costly to defend and divert
management’s attention from other aspects of our business.
Similarly, if the physicians or other providers or entities with
whom we do business are found to have violated abuse laws, they
may be subject to sanctions, which could also have a negative
impact on us.
Our
efforts to discover and develop potential products may not lead
to the discovery, development, commercialization or marketing of
actual drug products.
We are currently engaged in a number of different approaches to
discover and develop new product applications and product
candidates. At the present time, we have one Microcyn-based drug
candidate in clinical trials. We also have a non-Microcyn-based
compound in the research and development phase. We believe this
compound has potential applications in oncology. Discovery and
development of potential drug candidates are expensive and
time-consuming, and we do not know if our efforts will lead to
discovery of any drug candidates that can be successfully
developed and marketed. If our efforts do not lead to the
discovery of a suitable drug candidate, we may be unable to grow
our clinical pipeline or we may be unable to enter into
agreements with collaborators who are willing to develop our
drug candidates.
We
must implement additional and expensive finance and accounting
systems, procedures and controls as we grow our business and
organization and to satisfy new reporting requirements, which
will increase our costs and require additional management
resources.
As a public reporting company, we are required to comply with
the Sarbanes-Oxley Act of 2002 and the related rules and
regulations of the Securities and Exchange Commission, or the
Commission, including expanded disclosures and accelerated
reporting requirements and more complex accounting rules for the
reporting period ending March 31, 2008. Compliance with
Section 404 of the Sarbanes-Oxley Act of 2002 and other
requirements
42
will increase our costs and require additional management
resources. In a letter following their dismissal, our prior
independent auditors informed us that we did not have the
appropriate financial management and reporting structure in
place to meet the demands of a public company and that our
accounting and financial personnel lacked the appropriate level
of accounting knowledge, experience and training. Our current
independent auditors recommended certain changes in our internal
controls, which we are working on. We have upgraded our finance
and accounting systems, procedures and controls and will need to
continue to implement additional finance and accounting systems,
procedures and controls as we grow our business and
organization, enter into complex business transactions and take
actions designed to satisfy new reporting requirements.
Specifically, our experience with QP indicated that we need to
better plan for complex transactions and the application of
complex accounting principles relating to those transactions. If
we are unable to complete the required Section 404
assessment as to the adequacy of our internal control over
financial reporting, if we fail to maintain or implement
adequate controls, or if our independent registered public
accounting firm is unable to provide us with an unqualified
report as to the effectiveness of our internal control over
financial reporting as of the date of our second Annual Report
on
Form 10-K
for which compliance is required and thereafter, our ability to
obtain additional financing could be impaired. In addition,
investors could lose confidence in the reliability of our
internal control over financial reporting and in the accuracy of
our periodic reports filed under the Securities Exchange Act of
1934. A lack of investor confidence in the reliability and
accuracy of our public reporting could cause our stock price to
decline.
We may
not be able to maintain sufficient product liability insurance
to cover claims against us.
Product liability insurance for the healthcare industry is
generally expensive to the extent it is available at all. We may
not be able to maintain such insurance on acceptable terms or be
able to secure increased coverage if the commercialization of
our products progresses, nor can we be sure that existing or
future claims against us will be covered by our product
liability insurance. Moreover, the existing coverage of our
insurance policy or any rights of indemnification and
contribution that we may have may not be sufficient to offset
existing or future claims. A successful claim against us with
respect to uninsured liabilities or in excess of insurance
coverage and not subject to any indemnification or contribution
could have a material adverse effect on our future business,
financial condition, and results of operations.
Risks
Related to Our Common Stock
Our
operating results may fluctuate, which could cause our stock
price to decrease.
Fluctuations in our operating results may lead to fluctuations,
including declines, in our share price. Our operating results
and our share price may fluctuate from period to period due to a
variety of factors, including:
|
|
|
| |
•
|
demand by physicians, other medical staff and patients for our
Microcyn products;
|
| |
| |
•
|
reimbursement decisions by third-party payors and announcements
of those decisions;
|
| |
| |
•
|
clinical trial results and publication of results in
peer-reviewed journals or the presentation at medical
conferences;
|
| |
| |
•
|
the inclusion or exclusion of our Microcyn products in large
clinical trials conducted by others;
|
| |
| |
•
|
actual and anticipated fluctuations in our quarterly financial
and operating results;
|
| |
| |
•
|
developments or disputes concerning our intellectual property or
other proprietary rights;
|
| |
| |
•
|
issues in manufacturing our product candidates or products;
|
| |
| |
•
|
new or less expensive products and services or new technology
introduced or offered by our competitors or us;
|
| |
| |
•
|
the development and commercialization of product enhancements;
|
| |
| |
•
|
changes in the regulatory environment;
|
| |
| |
•
|
delays in establishing new strategic relationships;
|
43
|
|
|
| |
•
|
costs associated with collaborations and new product candidates;
|
| |
| |
•
|
introduction of technological innovations or new commercial
products by us or our competitors;
|
| |
| |
•
|
litigation or public concern about the safety of our product
candidates or products;
|
| |
| |
•
|
changes in recommendations of securities analysts or lack of
analyst coverage;
|
| |
| |
•
|
failure to meet analyst expectations regarding our operating
results;
|
| |
| |
•
|
additions or departures of key personnel; and
|
| |
| |
•
|
general market conditions.
|
Variations in the timing of our future revenues and expenses
could also cause significant fluctuations in our operating
results from period to period and may result in unanticipated
earning shortfalls or losses. In addition, the Nasdaq Global
Market, in general, and the market for life sciences companies,
in particular, have experienced significant price and volume
fluctuations that have often been unrelated or disproportionate
to the operating performance of those companies.
If an
active, liquid trading market for our common stock does not
develop, you may not be able to sell your shares quickly or at
or above the initial offering price.
Prior to our initial public offering, there was no public market
for our common stock. Although we listed our common stock listed
on the Nasdaq Global Market, an active and liquid trading market
for our common stock has not yet and may not ever develop or be
sustained. You may not be able to sell your shares quickly or at
or above the initial offering price if trading in our stock is
not active.
Future
sales of shares by our stockholders could cause the market price
of our common stock to drop significantly, even if our business
is doing well.
We had 11,844,411 outstanding shares of common stock-based on
the number of shares outstanding at March 31, 2007. The
outstanding shares will become available for resale in the
public market as shown in the chart below.
| |
|
|
|
Number of Restricted Shares
|
|
|
|
7,976,604 shares.
|
|
Date Available for Sale Into
Public Market Immediately upon expiration of the
180-day lock
up period
|
|
193,580 shares
|
|
At some point after expiration of
the 180-day
lock up period
|
Additional sales of our common stock in the public market, or
the perception that these sales could occur, could cause the
market price of our common stock to decline.
We do
not expect to pay dividends in the foreseeable future. As a
result, you must rely on stock appreciation, if any, for a
return on your investment.
We do not anticipate paying cash dividends on our common stock
in the foreseeable future. Any payment of cash dividends will
depend on our financial condition, results of operations,
capital requirements and other factors and will be at the
discretion of our Board of Directors. In addition, under two of
our secured loans, we will not pay any dividends without our
secured lenders’ prior written consent for as long as we
have any outstanding obligations to the secured lenders.
Accordingly, you will have to rely on appreciation in the price
of our common stock, if any, to earn a return on your investment
in our common stock. Furthermore, we may in the future become
subject to contractual restrictions on, or prohibitions against,
the payment of dividends.
44
We may
allocate our cash and cash equivalents in ways with which you
may not agree.
Our management has broad discretion in using our cash and cash
equivalents and may use them in ways with which you may
disagree. For example, we have deposited $2 million into a
segregated account to which we do not have access to assure
payment under one of our secured loan agreements. You and other
stockholders may not agree with our decisions about the use of
our reserves. Because we are not required to allocate our cash
and cash equivalents to any specific investment or transaction,
you cannot determine at this time the value or propriety of our
application of our cash position. Moreover, you will not have
the opportunity to evaluate the economic, financial or other
information on which we base our decisions on how to use our
cash and cash equivalents. As a result, we may use our cash and
cash equivalents for corporate purposes that do not immediately
enhance our prospects for the future or increase the value of
your investment.
Anti-takeover
provisions in our charter, by-laws and Delaware law may make it
more difficult for you to change our management and may also
make a takeover difficult.
Our corporate documents and Delaware law contain provisions that
limit the ability of stockholders to change our management and
may also enable our management to resist a takeover. These
provisions include:
|
|
|
| |
•
|
the ability of our Board of Directors to issue and designate the
rights of, without stockholder approval, up to
5,000,000 shares of convertible preferred stock, which
rights could be senior to those of common stock;
|
| |
| |
•
|
limitations on persons authorized to call a special meeting of
stockholders; and
|
| |
| |
•
|
advance notice procedures required for stockholders to make
nominations of candidates for election as directors or to bring
matters before an annual meeting of stockholders.
|
These provisions might discourage, delay or prevent a change of
control in our management. These provisions could also
discourage proxy contests and make it more difficult for you and
other stockholders to elect directors and cause us to take other
corporate actions. In addition, the existence of these
provisions, together with Delaware law, might hinder or delay an
attempted takeover other than through negotiations with our
Board of Directors.
Our
stockholders may experience substantial dilution in the value of
their investment if we issue additional shares of our capital
stock.
Our charter documents allow us to issue up to
100,000,000 shares of our common stock and to issue and
designate the rights of, without stockholder approval, up to
5,000,000 shares of convertible preferred stock. In the
event we issue additional shares of our capital stock, dilution
to our stockholders could result. In addition, if we issue and
designate a class of convertible preferred stock, these
securities may provide for rights, preferences or privileges
senior to those of holders of our common stock.
|
|
|
ITEM 1B:
|
Unresolved
Staff Comments
|
None.
We currently lease approximately 12,000 square feet of
office, research and manufacturing space in Petaluma,
California, which serves as our principal executive offices. We
also lease approximately 28,000 square feet of office space
in an adjacent building for manufacturing and research and
development. Both leases expire in September 2007. We are
currently in negotiations with our landlord and expect to extend
both leases.
We lease approximately 4,000 square feet of office space
and approximately 14,000 square feet of manufacturing and
warehouse space in Zapopan, Mexico, under a lease that expires
in April 2011. We lease approximately 5,000 square feet of
office space and approximately 14,000 square feet of
manufacturing and warehouse space in Sittard, The Netherlands,
under leases that expire in January 2009. As we expand, we may
need to establish manufacturing facilities in other countries.
45
We believe that, once we have determined whether to extend our
leases in Petaluma or to lease space elsewhere, our properties
will be adequate to meet our needs through March 2008.
|
|
|
ITEM 3.
|
Legal
Proceedings
|
In November 2005, the Company identified a possible criminal
misappropriation of its technology in Mexico, and it notified
the Mexican Attorney General’s office. The Company believes
the Mexican Attorney General is currently conducting an
investigation.
On March 14, 2006, the Company filed suit in the
U.S. District Court for the Northern District of California
against Nofil Corporation and Naoshi Kono, Chief Executive
Officer of Nofil for breach of contract, misappropriation of
trade secrets and trademark infringement. The Company believes
that Nofil Corporation violated key terms of both an exclusive
purchase agreement and non-disclosure agreement by contacting
and working with a potential competitor in Mexico. In the
complaint, the Company seeks damages of $3,500,000 and immediate
injunctive relief. On February 13, 2007, Nofil filed an
answer and cross-complaint, and it subsequently filed two
amendments to the cross complaint. The cross-complaint, which
alleges fraudulent inducement to enter contracts, breach of
non-disclosure contract, trade secret misappropriation,
conversion and violation under civil RICO statutes by the
Company, seeks damages of $4.5 million and equitable
relief. The Company believes that Nofil’s claims are
without merit and intends to defend its position with respect to
this matter. No trial date has been set.
The Company settled a trademark matter in Mexico in August 2006
asserting confusion in trademarks with respect to the
Company’s use of the name Microcyn60 in Mexico. Although
the Company believes that the nature and intended use of its
products are different from those with the similar names, it has
agreed with one of the parties to stop using the name Microcyn60
by September 2007. Company management believes that the name
change will satisfy an assertion that the Company’s
trademark use has caused confusion. Company management believes
that the Company could incur a possible loss of approximately
$100,000 for the use of the name Microcyn60 during the year
following the date of settlement.
On June 2, 2006, Oculus Technologies of México SA de
CV (OTM) filed an executory action in the Second Mercantile
Court at Guadalajara, Mexico, an action used to secure payment
of a commercial debt based on a promissory note signed by
Quimica Pasteur S de RL (QP) for approximately $770,000. The
attachment took place on February 7, 2007 and allowed OTM
to attach accounts receivables, bank accounts and some vehicles
of QP to secure the debt. No trial date has been set.
In June 2006, the Company received a written communication from
the grantor of a license to an earlier generation of its
technology indicating that such license was terminated due to an
alleged breach of the license agreement by the Company. The
license agreement extends to the Company’s use of the
technology in Japan only. While the Company does not believe
that the grantor’s revocation is valid under the terms of
the license agreement and no legal claim has been threatened to
date, the Company cannot provide any assurance that the grantor
will not take legal action to restrict the Company’s use of
the technology in the licensed territory.
While the Company management does not anticipate that the
outcome of this matter is likely to result in a material loss,
there can be no assurance that if the grantor pursues legal
action, such legal action would not have a material adverse
effect on the Company’s financial position or results of
operations.
In October 2004, we obtained EPA authorization, or registration,
for the distribution and sale of our Microcyn- based product as
a hospital grade disinfectant. In August 2006, we received a
“show cause” letter from the EPA stating that it was
prepared to file a civil administrative complaint against us for
violation of federal pesticide legislation in connection with
the sale or distribution of a pesticide that did not meet the
label’s efficacy claims. On April 5, 2007, we entered
into a Consent Agreement and Final Order with the EPA allowing
us to amend our EPA registration to a food sanitizer and pay a
$20,800 fine without admitting or denying any wrongdoing.
In April 2005, the Company was named as a defendant in an
employment related matter under a complaint filed by one of its
former employees in the Superior Court of the State of
California in the County of Sonoma. The Company entered into a
settlement agreement with the plaintiff in November 2006, which
provides for the payment of $250,000 cash and the issuance of a
warrant, valued at $365,000, to purchase 50,000 shares of
our common stock exercisable at $3.00 per share, and the
plaintiff has agreed to dismiss his claim and has waived any
other previous claims against us.
46
In September 2006, a consulting firm in Mexico City contacted
the Company threatening legal action in Mexico, alleging breach
of contract and claiming damages of $225,000. In December 2006,
the Company entered into a settlement agreement with the
consulting firm under which the Company paid $115,000 for the
dismissal of the consulting firm’s claim and release of all
claims against the Company.
The Company, on occasion, is involved in legal matters arising
in the ordinary course of its business. While management
believes that such matters are currently insignificant, there
can be no assurance that matters arising in the ordinary course
of business for which the Company is or could become involved in
litigation, will not have a material adverse effect on its
business, financial condition or results of operations.
|
|
|
ITEM 4.
|
Submission
of Matters to a Vote of Security Holders
|
None.
PART II
|
|
|
ITEM 5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities
|
(a) Our common stock is traded on the Nasdaq Global Market
under the symbol “OCLS” and has been trading since our
initial public offering on January 25, 2007. The following
table sets forth the range of high and low sale prices for our
common stock, based on the last daily sale, in each of the
quarters since our stock began trading:
| |
|
|
|
|
|
|
|
|
|
|
|
Low
|
|
|
High
|
|
|
|
|
2007:
|
|
|
|
|
|
|
|
|
|
Fourth Quarter (from
January 25, 2007)
|
|
$
|
5.66
|
|
|
$
|
8.40
|
|
According to the records of our transfer agent, we had 680
stockholders of record as of May 31, 2007.
(b) We have never declared or paid any cash dividends on
our capital stock, and we do not currently intend to pay any
cash dividends on our common stock in the foreseeable future.
Pursuant to our Loan and Security Agreement dated March 25,
2004 with Venture Lending & Leasing III, Inc. and
our Loan and Security Agreement dated June 14, 2006 with
Venture Lending & Leasing IV, Inc., each as amended,
we will not pay any dividends without our secured lenders’
prior written consent for so long as we have any outstanding
obligations to the secured lenders.
(c) Information regarding the securities authorized for
issuance under our equity compensation plans can be found under
Item 12 of Part III of this Report.
(d) On January 24, 2007, a Registration Statement on
Form S-1
(File
No. 333-135584)
relating to our initial public offering was declared effective
by the SEC. Roth Capital Partners acted as the managing
underwriter. The closing was January 30, 2007, and on
February 16, 2007, our underwriters exercised their option
to sell over-allotment shares. In total, the net offering
proceeds to us including over-allotment shares were
approximately $21.9 million (after deducting underwriting
discounts, commissions and offering expenses). Through
March 31, 2007, $3.7 million of the net proceeds were
used, including $845,000 for working capital and general
corporate purposes, $1.6 million for clinical trials and
related research and development, and $1.2 million for
sales and marketing activities worldwide. A portion of the net
proceeds may also be used to acquire or invest in complementary
businesses, technologies, services or products. Pending use for
these or other purposes, net proceeds have been invested in
interest bearing, investment grade securities.
(e) During the three months ended March 31, 2007, we
did not sell any equity securities that were not registered with
the Securities Act, nor did we purchase any of our equity
securities. However, during the fiscal year ended March 31,
2007 the period covered by this report, we had sales of
unregistered equity securities.
47
(f) Stock Performance Graph
The following information is not deemed to be “soliciting
material” or to be “filed” with the Securities
and Exchange Commission or subject to Regulation 14A or 14C
under the Securities Exchange Act of 1934 or to the liabilities
of Section 18 of the Securities Exchange Act of 1934, and
will not be deemed to be incorporated by reference into any
filing under the Securities Act of 1933 or the Securities
Exchange Act of 1934, except to the extent we specifically
incorporate it by reference into such a filing.
Set forth below is a line graph showing the cumulative total
stockholder return (change in stock price plus reinvested
dividends) assuming the investment of $100 on January 25,
2007 (the day of our initial public offering) in each of our
common stock, the NASDAQ Market Index and the NASDAQ
Biotechnology Index for the period commencing on
January 25, 2007 and ending on March 31, 2007. The
comparisons in the table are required by the Securities and
Exchange Commission and are not intended to forecast or be
indicative of future performance of our common stock.
COMPARISON
OF CUMULATIVE TOTAL RETURN
AMONG OCULUS INNOVATIVE SCIENCES, INC.,
NASDAQ COMPOSITE AND NASDAQ BIOTECH INDEX
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1/25/07
|
|
|
3/31/07
|
|
Oculus Innovative Sciences,
Inc.
|
|
|
$
|
100.00
|
|
|
|
$
|
76.28
|
|
|
NASDAQ Biotechnology Index
|
|
|
$
|
100.00
|
|
|
|
$
|
94.81
|
|
|
NASDAQ Composite Index
|
|
|
$
|
100.00
|
|
|
|
$
|
99.48
|
|
|
|
|
|
|
|
|
|
|
|
|
|
48
|
|
|
ITEM 6.
|
Selected
Financial Data
|
You should read the following selected consolidated financial
data together with “Management’s Discussion and
Analysis of Financial Condition and Results of Operations”
and our consolidated financial statements and related notes
included elsewhere in this
Form 10-K.
The selected consolidated statements of operations data for each
of the years ended March 31, 2007, 2006 and 2005 and the
selected consolidated balance sheet data as of March 31,
2007 and 2006 have been derived from our audited consolidated
financial statements and related notes included elsewhere in
this
Form 10-K.
The selected consolidated statements of operations data for the
years ended March 31, 2004 and 2003 and the selected
consolidated balance sheet data as of March 31, 2005, 2004
and 2003 have been derived from our consolidated financial
statements and related notes not included in this Annual Report
on
Form 10-K.
The selected consolidated statement of operations data for the
year ended March 31, 2003 and the selected consolidated
balance sheet data as of March 31, 2003 have not been
audited. The unaudited financial statements include, in the
opinion of management, all adjustments that management considers
necessary for the fair presentation of the financial information
set forth in those statements. Our historical results are not
necessarily indicative of the results that may be expected in
the future.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
2004
|
|
|
2003
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Unaudited)
|
|
|
|
|
Consolidated statement of
operations data (in thousands, except per share data):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product
|
|
$
|
3,679
|
|
|
$
|
1,966
|
|
|
$
|
473
|
|
|
$
|
95
|
|
|
$
|
—
|
|
|
Service
|
|
|
864
|
|
|
|
618
|
|
|
|
883
|
|
|
|
807
|
|
|
|
2,470
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
4,543
|
|
|
|
2,584
|
|
|
|
1,356
|
|
|
|
902
|
|
|
|
2,470
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product(1)
|
|
|
2,104
|
|
|
|
3,899
|
|
|
|
2,211
|
|
|
|
1,403
|
|
|
|
—
|
|
|
Service(1)
|
|
|
895
|
|
|
|
1,003
|
|
|
|
1,311
|
|
|
|
1,265
|
|
|
|
1,768
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total cost of revenues
|
|
|
2,999
|
|
|
|
4,902
|
|
|
|
3,522
|
|
|
|
2,668
|
|
|
|
1,768
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit (loss)
|
|
|
1,544
|
|
|
|
(2,318
|
)
|
|
|
(2,166
|
)
|
|
|
(1,766
|
)
|
|
|
702
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development(1)
|
|
|
4,508
|
|
|
|
2,600
|
|
|
|
1,654
|
|
|
|
1,413
|
|
|
|
68
|
|
|
Selling, general and
administrative(1)
|
|
|
16,520
|
|
|
|
15,933
|
|
|
|
12,492
|
|
|
|
3,918
|
|
|
|
2,102
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
21,028
|
|
|
|
18,533
|
|
|
|
14,146
|
|
|
|
5,331
|
|
|
|
2,170
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(19,484
|
)
|
|
|
(20,851
|
)
|
|
|
(16,312
|
)
|
|
|
(7,097
|
)
|
|
|
(1,468
|
)
|
|
Interest expense
|
|
|
(956
|
)
|
|
|
(172
|
)
|
|
|
(372
|
)
|
|
|
(178
|
)
|
|
|
(123
|
)
|
|
Interest income
|
|
|
312
|
|
|
|
282
|
|
|
|
8
|
|
|
|
3
|
|
|
|
—
|
|
|
Other income (expense), net
|
|
|
345
|
|
|
|
(377
|
)
|
|
|
146
|
|
|
|
(26
|
)
|
|
|
(4
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss from continuing operations
|
|
|
(19,783
|
)
|
|
|
(21,118
|
)
|
|
|
(16,530
|
)
|
|
|
(7,298
|
)
|
|
|
(1,595
|
)
|
|
Loss from operations of
discontinued business
|
|
|
—
|
|
|
|
(818
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Loss on disposal of discontinued
business
|
|
|
—
|
|
|
|
(1,163
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss on discontinued operations
|
|
|
—
|
|
|
|
(1,981
|
)
|
|
|
—
|
|
|
|
(7,298
|
)
|
|
|
(1,595
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(19,783
|
)
|
|
|
(23,099
|
)
|
|
|
(16,530
|
)
|
|
|
(7,298
|
)
|
|
|
(1,595
|
)
|
|
Preferred stock dividends
|
|
|
(404
|
)
|
|
|
(121
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss available to common
stockholders
|
|
$
|
(20,187
|
)
|
|
$
|
(23,220
|
)
|
|
$
|
(16,530
|
)
|
|
$
|
(7,298
|
)
|
|
$
|
(1,595
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share: basic
and diluted
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Continuing operations
|
|
$
|
(3.71
|
)
|
|
$
|
(5.12
|
)
|
|
$
|
(4.22
|
)
|
|
$
|
(1.87
|
)
|
|
$
|
(0.42
|
)
|
|
Discontinued operations
|
|
|
—
|
|
|
|
(0.48
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
(3.71
|
)
|
|
$
|
(5.60
|
)
|
|
$
|
(4.22
|
)
|
|
$
|
(1.87
|
)
|
|
$
|
(0.42
|
)
|
|
Weighted-average number of shares
used in per common share calculations:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted
|
|
|
5,448
|
|
|
|
4,150
|
|
|
|
3,914
|
|
|
|
3,911
|
|
|
|
3,827
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Includes the following stock-based compensation charges (in
thousands): |
49
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
2004
|
|
|
2003
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Unaudited)
|
|
|
|
|
Cost of revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product
|
|
$
|
—
|
|
|
$
|
2
|
|
|
$
|
2
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Service
|
|
|
4
|
|
|
|
1
|
|
|
|
3
|
|
|
|
10
|
|
|
|
55
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
70
|
|
|
|
52
|
|
|
|
5
|
|
|
|
56
|
|
|
|
—
|
|
|
Selling, general and administrative
|
|
|
1,508
|
|
|
|
542
|
|
|
|
2,339
|
|
|
|
358
|
|
|
|
186
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
2004
|
|
|
2003
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(Unaudited)
|
|
|
|
|
Consolidated Balance Sheet Data
(in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
19,050
|
|
|
$
|
7,448
|
|
|
$
|
3,287
|
|
|
$
|
869
|
|
|
$
|
177
|
|
|
Working capital
|
|
|
13,834
|
|
|
|
5,127
|
|
|
|
663
|
|
|
|
(1,186
|
)
|
|
|
(145
|
)
|
|
Total assets
|
|
|
26,9
|
|
|
|
12,689
|
|
|
|
6,940
|
|
|
|
2,992
|
|
|
|
961
|
|
|
Total liabilities
|
|
|
12,049
|
|
|
|
5,351
|
|
|
|
4,738
|
|
|
|
3,374
|
|
|
|
1,040
|
|
|
Total stockholders’ equity
(deficit)
|
|
|
14,901
|
|
|
|
7,338
|
|
|
|
2,202
|
|
|
|
(382
|
)
|
|
|
(79
|
)
|
|
|
|
ITEM 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations
Business
Overview
Oculus Innovative Sciences is a biopharmaceutical company that
develops, manufactures and markets a family of products, based
on its platform technology called Microcyn, intended to help
prevent and treat infections in chronic and acute wounds.
Microcyn is a non-irritating oxychlorine compound designed to
treat a wide range of pathogens, including antibiotic-resistant
strains of bacteria, viruses, fungi and spores.
Financial
Operations Overview
Revenues
We derive our revenues from product sales and service
arrangements. Product revenues are generated from the sale of
Microcyn products to hospitals, medical centers, doctors,
pharmacies, distributors and strategic partners, and are
generally recorded upon shipment following receipt of a purchase
order or upon obtaining proof of sell-through by a distributor.
Product sales are made either through direct sales personnel or
distributors.
Service revenues are derived from consulting and testing
contracts. Service revenues are generally recorded upon
performance under the service contract. Revenues generated from
testing contracts are recorded upon completion of the test and
when the final report is sent to the customer. We have refocused
our business efforts away from consulting and testing services
toward the completion of our clinical trials and eventual
commercialization of Microcyn. As a result, we expect service
revenues to continue to significantly decline in future periods.
Cost of
Revenues
Cost of product revenues represents the costs associated with
the manufacturing of our products, including expenses for our
various facilities which are fixed, and related personnel cost
and the cost of materials used to produce our products. Cost of
service revenues consists primarily of personnel related
expenses and supplies.
50
Research
and Development Expense
Research and development expense consists of costs related to
the research and development of Microcyn and our manufacturing
process, the development of new products and new delivery
systems for our products and to carry out preclinical studies
and clinical trials to obtain various regulatory approvals.
Research and development expense is charged as incurred.
Selling,
General and Administrative Expense
Selling, general and administrative expense consists of
personnel related costs, including salaries and sales
commissions, and education and promotional expenses associated
with Microcyn and costs related to administrative personnel and
senior management. These expenses also include the costs of
educating physicians and other healthcare professionals
regarding our products and participating in industry conferences
and seminars. Selling, general and administrative expense also
includes travel costs, outside consulting services, legal and
accounting fees and other professional and administrative costs.
Stock-Based
Compensation Expense
Prior to April 1, 2006, we accounted for stock-based
employee compensation arrangements in accordance with the
provisions of APB No. 25, “Accounting for Stock Issued
to Employees,” and its related interpretations and applied
the disclosure requirements of SFAS No. 148,
“Accounting for Stock-Based Compensation-Transition and
Disclosure, an amendment of FASB Statement No. 123.”
We used the minimum value method to measure the fair value of
awards issued prior to April 1, 2006 with respect to its
application of the disclosure requirements under SFAS 123.
Effective April 1, 2006, we adopted
SFAS No. 123(R) “Share Based Payment”
(“SFAS 123(R)”). This statement is a revision of
SFAS Statement No. 123, and supersedes APB Opinion
No. 25, and its related implementation guidance.
SFAS 123(R) addresses all forms of share based payment
(“SBP”) awards including shares issued under employee
stock purchase plans, stock options, restricted stock and stock
appreciation rights. Under SFAS 123(R), SBP awards result
in a cost that will be measured at fair value on the
awards’ grant date, based on the estimated number of awards
that are expected to vest and will result in a charge to
operations.
We had a choice of two attribution methods for allocating
compensation costs under SFAS 123(R): the
“straight-line method,” which allocates expense on a
straight-line basis over the requisite service period of the
last separately vesting portion of an award, or the “graded
vesting attribution method,” which allocates expense on a
straight-line basis over the requisite service period for each
separately vesting portion of the award as if the award was, in
substance, multiple awards. We chose the former method and
amortized the fair value of each option on a straight-line basis
over the requisite period of the last separately vesting portion
of each award.
Under SFAS 123(R), nonpublic entities, including those that
become public entities after June 15, 2005, that used the
minimum value method of measuring equity share options and
similar instruments for either recognition or pro forma
disclosure purposes under Statement 123 are required to
apply SFAS 123(R)prospectively to new awards and to awards
modified, repurchased, or cancelled after the date of adoption.
In addition, SFAS 123(R), requires such entities to
continue accounting for any portion of awards outstanding at the
date of initial application using the accounting principles
originally applied to those awards. Accordingly, stock-based
compensation expense relating to awards granted prior to
April 1, 2006 that are expected to vest in periods ending
after April 1, 2006 are being recorded in accordance with
the provisions of APB 25 and its related interpretive
guidance.
We have adopted the prospective method with respect to
accounting for its transition to SFAS 123(R). Accordingly,
we recognized in salaries and related expense in the statement
of operations $158,000 of stock-based compensation expense
during the year ended March 31, 2007, which represents the
intrinsic value amortization of options granted prior to
April 1, 2006 that we are continuing to account for using
the recognition and measurement principles prescribed under
APB 25. We also recognized in salaries and related expense
in the statement of operations $815,000 of stock-based
compensation expense during the year ended March 31, 2007
which represents the amortization of the fair value of options
granted subsequent to adoption of SFAS 123(R). During the
year ended March 31, 2007, we reclassified certain
components of our stockholders’ equity section to reflect
the elimination of
51
deferred compensation arising from unvested share-based
compensation pursuant to the requirements of Staff Accounting
Bulletin No. 107, regarding Statement of Financial
Accounting Standards No. 123(R), “Share-Based
Payment.” This deferred compensation was previously
recorded as an increase to additional paid-in capital with a
corresponding reduction to stockholders’ equity for such
deferred compensation. This reclassification has no effect on
net income or total stockholders’ equity as previously
reported. We will record an increase to additional paid-in
capital as the share-based payments vest.
Total non-cash stock-compensation charges were
$1.6 million, $597,000, and $2.3 million for the years
ended March 31, 2007, 2006, and 2005 respectively. The
losses from operations excluding non-cash stock-compensation
charges were $17.9 million, $20.3 million, and
$14.0 million for the years ended March 31, 2007,
2006, and 2005 respectively.
Comparison
of Years Ended March 31, 2007 and 2006
Revenues
We experienced growth in revenues in both our product and
services businesses resulting in reported revenues of
$4.5 million in the year ended March 31, 2007, an
increase of 76% from the prior year level of $2.6 million.
The $1.7 million, or 87%, increase in product revenues was
due primarily to higher sales volumes in Mexico and Europe, and
sales to a new customer, Alkem Laboratories Limited, in India.
The following table shows our product revenues by country (in
thousands); note that sales in India are reported as part of our
Europe business:
| |
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
(In thousands)
|
|
|
|
|
U.S
|
|
$
|
140
|
|
|
$
|
109
|
|
|
Mexico
|
|
|
2,513
|
|
|
|
1,788
|
|
|
India
|
|
|
604
|
|
|
|
—
|
|
|
Europe
|
|
|
422
|
|
|
|
69
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
3,679
|
|
|
$
|
1,966
|
|
|
|
|
|
|
|
|
|
|
|
The $246,000, or 40%, increase in service revenues was due
primarily to an increase in the volume of tests provided by our
services business. We expect that our service revenues will
significantly decline in future periods, as we continue to
implement our strategy of focusing primarily on our Microcyn
business.
Gross
Profit/Loss
We reported gross profit from our Microcyn products business of
$1.6 million, or 43% of product revenues, in the year ended
March 31, 2007, compared to the prior year reported gross
loss of $1.9 million, or -98% of product revenues. The
gross loss in the year ended March 31, 2006 was adversely
affected by $1.0 million of non-recurring inventory
write-downs and approximately $200,000 of non-recurring charges
associated with the relocation of our Mexico manufacturing
facility during the year. Excluding these charges, our gross
loss from product sales in the year ended March 31, 2006
would have been $685,000, or -35% of product revenues. The
increase in product gross margin, excluding these non-recurring
charges, from -35% in the year ended March 31, 2006, to 43%
in the year ended March 31, 2007, was primarily due to
improvements in manufacturing efficiencies through the
consolidation of our worldwide manufacturing from three sites to
two during the year. In April 2006, we transitioned the United
States facility from a product manufacturing site into a
research and development facility, and began producing all
products for sales outside of Mexico in our facility in Europe.
The remaining improvements in gross profits were due to a full
year of benefit from the relocation of our Mexican manufacturing
site during the year ended March 31, 2006 from Morelia,
Mexico into a lower-cost manufacturing facility in Zapopan,
Mexico.
We reported a gross loss from our services business of $31,000,
or -4% of service revenues, in the year ended March 31,
2007, compared to the prior year reported gross loss of
$385,000, or -62% of service revenues. This improvement in
margin from -62% to -4% was due primarily to the growth in sales
volume of our services which
52
moved our service revenues closer to exceeding the relatively
high fixed cost of our laboratory facility. Additionally, we
discontinued our consulting service business during the year
ended March 31, 2006 which had a positive impact on our
services margin.
We expect gross profits to increase as a percentage of sales in
future periods as we continue to implement our strategy of
focusing on our Microcyn business, and we move away from our
lower-margin services business.
Research
and Development Expense
Research and development expense increased $1.9 million, or
73%, to $4.5 million for the year ended March 31,
2007, from $2.6 million for the year ended March 31,
2006. This increase was primarily the result of higher personnel
costs associated with the expansion of our research and
development teams. The expansion of these teams was through both
an internal shift of focus in our United States operations from
product manufacturing to research and development, as well as
through the hiring of outside personnel. The expansion of the
research and development teams helps support our increased
attention to product development and the management of
regulatory trials designed to obtain FDA drug approvals for our
Microcyn products.
We expect research and development expenses to increase
significantly in future periods as we incur the costs associated
with our FDA trials for the treatment of diabetic foot ulcer
infections with Dermacyn, and as we expand the scope of our new
product development.
Selling,
General and Administrative Expense
Selling, general and administrative expense increased $587,000,
or 4%, to $16.5 million for the year ended March 31,
2007, from $15.9 million for the year ended March 31,
2006. This increase was primarily due to a $966,000 increase in
non-cash stock-based compensation expense recorded during the
year ended March 31, 2007. This increase in stock
compensation charges was primarily the result of charges
incurred for the warrants issued to a new board member, which
are amortized over the two-year agreement, and for the warrants
issued for the settlement of litigation with a past employee.
This increase was offset in the current year by lower outside
service expenses, primarily in legal and accounting fees.
We expect that general and administrative costs will increase in
future periods due to the added expenses of being a public
company, including insurance, legal, accounting, and fees
associated with Sarbanes Oxley compliance. These increases may
be offset in part by lower selling expenses as we shift our
company resources from the opening of new markets worldwide to
the completion of our FDA regulatory trials, and as we expand
the scope of our new product development.
Interest
income and expense and other income and expense
Interest expense increased $784,000 to $956,000 for the year
ended March 31, 2007, from $172,000 in the year ended
March 31, 2006, primarily due to the issuance of
$8.2 million of new debt during the year. The new debt had
amortized debt issuance costs booked as non-cash interest of
$402,000, and normal interest expense of $441,000 during the
year ended March 31, 2007.
Other income and expense primarily consists of non-cash charges
due to the fluctuation of foreign exchange rates, and the
resulting gain or loss booked for the revaluation of our
intercompany notes payable denominated in non-local currencies.
During the year ended March 31, 2007, the U.S. dollar
became stronger in relation to the Mexican peso and the Euro,
and a net $407,000 gain on foreign exchange was recorded
accordingly. In comparison, during the year ended March 31,
2006, the U.S. Dollar became weaker in relation to the
Mexican Peso and the Euro, and a $283,000 loss on foreign
exchange was recorded. Additionally, during the year ended
March 31, 2006, we incurred $113,000 in loss on the
disposal of assets.
Due to the difficulty of predicting foreign currency
fluctuations, we do not know the affect that such fluctuations
may have on our operating results in future periods.
53
Comparison
of Years Ended March 31, 2006 and March 31,
2005
Revenues
Revenues increased $1.2 million, or 91%, to
$2.6 million for the year ended March 31, 2006, from
$1.4 million for the year ended March 31, 2005.
Product revenues increased $1.5 million, or 316%, to
$2.0 million for the year ended March 31, 2006, from
$473,000 for the year ended March 31, 2005. This increase
was primarily due to a $1.4 million increase in sales of
Microcyn60 in Mexico following the expansion of our sales force
in that country and the receipt of product reimbursement by the
Mexican Ministry of Health.
The increase in product revenues was partially offset by a
$265,000 decrease in service revenues during the year ended
March 31, 2006, as compared to the prior year. The decrease
in service revenues was a result of a shift in our focus from
services to the development of our Microcyn products during the
year ended March 31, 2006.
Gross
Profit/Loss
We reported a gross loss from our Microcyn products business of
$1.9 million, or -98% of revenues, in the year ended
March 31, 2006, compared to the prior year reported gross
loss of $1.7 million, or -367% of product revenues. The
gross loss in the year ended March 31, 2006 was adversely
affected by $1.0 million of non-recurring inventory
write-downs and approximately $200,000 of non-recurring charges
associated with the relocation of our Mexico manufacturing
facility during the year. Excluding these charges, our gross
loss from product sales in the year ended March 31, 2006
would have been $685,000, or -35% of product revenues. The
increase in margin, excluding these non-recurring charges, from
-367% in the year ended March 31, 2005, to -35% in the year
ended March 31, 2006, was primarily due to the growth in
sales volume which moved our product revenues closer to
exceeding our relatively high fixed cost of manufacturing. The
remaining improvements in gross profits were due to the
relocation of our Mexican manufacturing site during the year
ended March 31, 2006 from Morelia, Mexico into a lower-cost
manufacturing facility in Zapopan, Mexico.
We reported a gross loss from our services business of $385,000,
or -62% of service revenues, during the year ended
March 31, 2007, compared to the prior year reported gross
loss of $428,000, or -48% of service revenues. This decrease in
margin from -48% to -62% was due primarily to the decline in
sales volume of our services which moved our service revenues
further away from exceeding the relatively high fixed cost of
our laboratory facility.
Research
and Development Expense
Research and development expense increased $946,000, or 57%, to
$2.6 million during the year ended March 31, 2006,
from $1.7 million in the year ended March 31, 2005.
This increase was primarily attributable to the expansion of our
regulatory team, which focused on FDA and KEMA approvals for
Microcyn products during the period. Additionally, in September
2005, we commenced our pre-operative skin preparation pilot
studies to support our application for an FDA drug clearance
indicating microbial load reduction. Total spending on
regulatory trials, other clinical studies, and related expenses
increased $1.2 million, or 164%, to $1.9 million for
the year ended March 31, 2006, from $735,000 during the
year ended March 31, 2005. This increase was partially
offset by a $418,000 decrease in spending on new product
development to $497,000 in the year ended March 31, 2006,
from $915,000 in the year ended March 31, 2005.
Selling,
General and Administrative Expense
Selling, general and administrative expense increased
$3.4 million, or 28%, to $15.9 million during the year
ended March 31, 2006, from $12.5 million during the
year ended March 31, 2005. This increase was partially due
to a $1.5 million increase in United States selling,
general and administrative expense primarily as a result of
higher outside consulting and service fees during the year ended
March 31, 2006. Specifically, outside accounting fees
increased by $653,000 due to the preparation and completion of
an audit of our last four fiscal years, legal fees increased by
$507,000 due to expanded intellectual property and general legal
support, and outside consulting and service fees increased by
$294,000 due to consulting expenses related to the marketing of
our products in Asia.
In addition, sales and marketing expense in Europe increased
$429,000 due to the hiring of additional sales and marketing
personnel during the year ended March 31, 2006.
54
Selling, general and administrative expense in Mexico increased
$3.3 million during the year ended March 31, 2006
compared to the prior year primarily due to expanded sales and
marketing efforts in Mexico, as well as non-recurring charges
associated with the relocation of our Mexican subsidiary’s
facility. During the year ended March 31, 2006, we began
utilizing 75 full-time, direct sales personnel in the major
districts of Mexico, dedicated to the sale of Microcyn60 in the
hospital and pharmacy markets in Mexico. As a result, sales and
marketing expense in Mexico increased $2.7 million during
the year ended March 31, 2006, compared to the prior year.
The increase in selling, general and administrative expense was
offset by a $1.8 million decrease in non-cash stock
compensation expense in the year ended March 31, 2006
compared to the prior year. Approximately $1.7 million of
non-cash stock-based compensation expense incurred during the
year ended March 31, 2005 was related to the grant of an
option to purchase 300,000 shares of common stock to our
Chief Executive Officer.
Interest
income and expense and other income and expense
Interest expense decreased $200,000 to $172,000 for the year
ended March 31, 2006, from $372,000 during the year ended
March 31, 2005. This decrease was primarily the result of
lower average borrowings during the year as our outstanding debt
was paid down. Interest income increased $274,000, to $282,000
during the year ended March 31, 2006, from $8,000 in the
year ended March 31, 2005. This increase was primarily the
result of higher balances of interest-bearing instruments during
the year ended March 31, 2006.
Other income and expense primarily consists of non-cash charges
due to the fluctuation of foreign exchange rates, and the
resulting gain or loss booked for the revaluation of our
intercompany notes payable denominated in non-local currencies.
During the year ended March 31, 2006 the U.S. dollar
became weaker in relation to the Mexican peso and the Euro, and
a $283,000 loss on foreign exchange was booked accordingly. In
comparison, during the year ended March 31, 2005, the
U.S. dollar became stronger in relation to the Mexican Peso
and the Euro, and a $134,000 gain on foreign exchange was
booked. Additionally, during the year ended March 31, 2006,
Oculus incurred $113,000 in loss on the disposal of assets.
Discontinued
Operations
Loss on discontinued operations was $2.0 million during the
year ended March 31, 2006. This loss consisted of $818,000
classified as a loss from operations of discontinued business
and $1.2 million of loss on the disposal of discontinued
business. The loss from operations of discontinued business
represents the net operating loss of QP, which was consolidated
with our financial results. The relationship was terminated in
the fourth quarter of the year ended March 31, 2006 and the
loss was classified as a discontinued operation on our
consolidated statements of operations. As no relationship
existed with this entity prior to the year ended March 31,
2006, no charges were recognized in prior years.
Liquidity
and Capital Resources
Since our inception, we have incurred significant losses and, as
of March 31, 2007, we had an accumulated deficit of
approximately $70.5 million. We have not yet achieved
profitability, and we expect that our operating expenses will
continue to increase and, as a result, we will need to generate
significant product revenues to achieve profitability in the
future.
Sources
of Liquidity
Since our inception, substantially all of our operations have
been financed through the sale of our common and convertible
preferred stock. Through March 31, 2007, we had received
net proceeds of $21.9 million through the sale of our
common stock in our initial public offering in January 2007,
$3.5 million from prior sales of common stock,
$6.7 million from the sale of Series A convertible
preferred stock, $43.7 million from the sale of
Series B convertible preferred stock, $2.9 million
from the sales of Series C convertible preferred stock, and
$304,000 from the issuance of common stock to employees,
consultants and directors in connection with the exercise of
stock options. We have received additional funding through
various debt and financing transactions, as described below. We
have also used our revenues to date as a source of additional
liquidity. As of March 31, 2007, we had unrestricted
55
cash and cash equivalents of $19.1 million, and restricted
cash of $2.0 million reserved for the pay-down of
particular debt.
In June 2006, we entered into a loan and security agreement with
a financial institution to borrow a maximum of
$5.0 million. Under this facility we have borrowed
$4.2 million, and have paid back $852,000 in principle as
of March 31, 2007. The terms of this facility include
monthly principal repayment over three years, and an annual
interest rate of 8.5%. In conjunction with this agreement, we
issued warrants to purchase up to 75,000 shares of our
Series B preferred stock at an exercise price of
$18.00 per share. Warrants to purchase 53,750 shares
were earned and exercisable at execution of the agreement.
On November 7, 2006, we signed a loan agreement with Robert
Burlingame, one of the Company’s directors, under which
Mr. Burlingame advanced to us $4.0 million, which was
funded on November 10, 2006, and which accrues interest at
an annual interest rate of 7%. The principal and all accrued
interest under the loan agreement will become due and payable in
full on November 10, 2007. The loan is secured by all of
our assets, other than our intellectual property, but is
subordinate to the security interest held by our secured lender.
Brookstreet Securities Corporation, a placement agent, was paid
a fee in the amount of $50,000 and granted a warrant to purchase
25,000 shares of our common stock at an exercise price of
$18.00 per share in connection with this loan.
Additionally, in May 2007, we deposited $2 million into a
segregated, interest-bearing account on which
Mr. Burlingame, has sole signature authority. Under our
arrangement with Mr. Burlingame, the interest on the
account is paid to us each month, and the $2 million
principal will be used to satisfy a part of our obligation to
Mr. Burlingame under the loan agreement. We have also
agreed to deposit an additional $2 million into this
segregated account should our total cash and restricted cash
balances drop to below $10 million in total.
Cash
Flows
As of March 31, 2007, we had unrestricted cash and cash
equivalents of $19.1 million, compared to $7.4 million
at March 31, 2006 and $3.3 million at March 31,
2005.
Net cash used in operating activities was $18.1 million,
$19.7 million and $13.5 million during the years ended
March 31, 2007, 2006 and 2005, respectively. Net cash used
in each of these periods primarily reflects net loss for these
periods, offset in part by non-cash charges in operating assets
and liabilities, non-cash stock-based compensation and
depreciation and amortization.
Net cash used in investing activities was $877,000, $419,000 and
$1.1 million for the years ended March 31, 2007, 2006
and 2005, respectively. Primarily this cash was used to invest
in fixed assets and other capital expenditures to support
increased personnel and manufacturing facility expansion in
Europe and Mexico during the years ended March 31, 2007,
2006 and 2005.
Net cash provided by financing activities was
$30.6 million, $26.1 million and $17.2 million
for the years ended March 31, 2007, 2006 and 2005,
respectively. The net cash provided by financing activities for
the year ended March 31, 2007 was primarily due to our
initial public offering, which raised net cash of
$21.9 million. This amount includes $24.2 million of
net cash raised in our initial public offering on
January 30, 2007 (net of underwriter’s commissions,
and expenses), and $2.4 million of net cash raised when our
underwriters exercised their option to sell over allotment
shares on February 16, 2007. These amounts were offset by
$2.8 million of additional accounting, legal, and other
expenses directly related to the initial public offering.
Additionally, the sale of convertible preferred stock, generated
$2.9 million, $27.0 million and $16.7 million for
the years ended March 31, 2007, 2006 and 2005,
respectively. In addition, net proceeds from debt financing
added $7.2 million and $541,000 during the years ended
March 31, 2007 and 2005, respectively, and net payments on
debt were $696,000 during the year ended March 31, 2006.
During the year ended March 31, 2007, the activity was also
related to the $2.0 million reclassification of cash to
restricted cash per the terms of the Burlingame loan agreement,
as described above.
56
Contractual
Obligations
As of March 31, 2007, we had contractual obligations as
follows (long-term debt and capital lease amounts include
principal payments only):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
|
|
|
Less Than
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
1 Year
|
|
|
1-3 Years
|
|
|
4-5 Years
|
|
|
After 5 Years
|
|
|
|
|
Long-term debt
|
|
$
|
8,035
|
|
|
$
|
6,045
|
|
|
$
|
1,952
|
|
|
$
|
23
|
|
|
$
|
15
|
|
|
Capital leases
|
|
|
42
|
|
|
|
17
|
|
|
|
25
|
|
|
|
—
|
|
|
|
—
|
|
|
Operating leases
|
|
|
660
|
|
|
|
296
|
|
|
|
261
|
|
|
|
103
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
8,737
|
|
|
$
|
6,358
|
|
|
$
|
2,238
|
|
|
$
|
126
|
|
|
$
|
15
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
We have operating leases covering approximately
40,000 square feet of office and manufacturing space in
Petaluma, California, expiring in September 2007, and our
monthly rent is $23,493. We also have operating leases covering
approximately 19,000 square feet of office and
manufacturing space in Sittard, The Netherlands expiring in
2009, and approximately 12,000 square feet of office and
manufacturing space and 5,000 square feet of warehouse
space in Zapopan, Mexico, expiring in 2011 and 2007,
respectively.
We do not have any off-balance sheet arrangements as such term
is defined in rules promulgated by the SEC.
Operating
Capital and Capital Expenditure Requirements
We expect to continue to incur substantial operating losses in
the future as we continue our FDA clinical trials on our
Microcyn technology to treat diabetic foot ulcers, and the
subsequent commercialization of an FDA approved drug. It may
take several years to obtain the necessary regulatory approvals
to commercialize Microcyn as a drug in the United States.
We currently anticipate that our cash, cash equivalents, and
restricted cash balances, together with our future revenues and
interest we earn on these balances will be sufficient to meet
our anticipated cash requirements through March 31, 2008.
We currently anticipate a need to raise additional funds prior
to March 31, 2008, and in periods following, through public
or private equity offerings, debt financings, corporate
collaborations or other means in order to complete our
Phase 3 clinical trials, execute our product development
strategy, and to commercialize Microcyn as a drug product in the
United States. The sale of additional equity or convertible debt
securities could result in dilution to our stockholders. To the
extent that we raise additional funds through collaborative
arrangements, it may be necessary to relinquish some rights to
our technologies or grant licenses on terms that are not
favorable to us. We do not know whether additional funding will
be available on acceptable terms, or at all. A failure to secure
additional funding when needed may require us to curtail certain
operational activities, including regulatory trials, sales and
marketing, and international operations and would have a
material adverse effect on our future business and financial
condition.
Our future funding requirements will depend on many factors,
including:
|
|
|
| |
•
|
the scope, rate of progress and cost of our clinical trials and
other research and development activities;
|
| |
| |
•
|
future clinical trial results;
|
| |
| |
•
|
the terms and timing of any collaborative, licensing and other
arrangements that we may establish;
|
| |
| |
•
|
the cost and timing of regulatory approvals;
|
| |
| |
•
|
the cost and delays in product development as a result of any
changes in regulatory oversight applicable to our products;
|
| |
| |
•
|
the cost and timing of establishing sales, marketing and
distribution capabilities;
|
| |
| |
•
|
the effect of competing technological and market developments;
|
57
|
|
|
| |
•
|
the cost of filing, prosecuting, defending and enforcing any
patent claims and other intellectual property rights; and
|
| |
| |
•
|
the extent to which we acquire or invest in businesses, products
and technologies.
|
Use of
Estimates
The preparation of consolidated financial statements in
conformity with United States generally accepted accounting
principles requires management to make estimates and assumptions
that affect the reported amounts of assets and liabilities and
disclosures of contingent liabilities at the dates of the
consolidated financial statements and the reported amounts of
revenues and expenses during the reporting periods. Actual
results could differ from these estimates. These estimates and
assumptions include reserves and write-downs related to
receivables and inventories, the recoverability of long-term
assets, deferred taxes and related valuation allowances and
valuation of equity instruments.
Recent
Accounting Pronouncements
In March 2006, the FASB issued SFAS No. 156
“Accounting for Servicing of Financial Assets —
an amendment of FASB Statement No. 140”
(“SFAS 156”). SFAS 156 is effective for the
first fiscal year beginning after September 15, 2006.
SFAS 156 changes the way entities account for servicing
assets and obligations associated with financial assets acquired
or disposed of. We have not yet completed our evaluation of the
impact of adopting SFAS 156 on our results of consolidated
operations or financial position, and do not expect that the
adoption of SFAS 156 will have a material impact on our
consolidated financial statements.
In June 2006, the Financial Accounting Standards Board issued
FASB Interpretation No. 48, Accounting for Uncertainty in
Income Taxes. This Interpretation prescribes a recognition
threshold and measurement attribute for the financial statement
recognition and measurement of a tax position taken or expected
to be taken in a tax return. This Interpretation also provides
guidance on derecognition, classification, interest and
penalties, accounting in interim periods, disclosure, and
transition. The Interpretation is effective for fiscal years
beginning after December 15, 2006. The Company has not yet
completed its analysis of the impact this Interpretation will
have on its financial condition, results of operations, cash
flows or disclosures.
In February 2007, the FASB issued SFAS 159, “The Fair
Value Option for Financial Assets and Financial
Liabilities” (“SFAS 159”). SFAS 159
provides companies with an option to report selected financial
assets and liabilities at fair value and establishes
presentation and disclosure requirements designed to facilitate
comparisons between companies that choose different measurement
attributes for similar types of assets and liabilities.
SFAS 159 is effective for fiscal years beginning after
November 15, 2007. The Company is in the process of
evaluating the impact of the adoption of this statement on the
Company’s results of operations and financial condition.
In September 2006, the FASB issued SFAS No. 157,
“Accounting for Fair Value Measurements”
(“SFAS 157”). SFAS 157 defines fair value,
and establishes a framework for measuring fair value in
generally accepted accounting principles and expands disclosure
about fair value measurements. SFAS 157 is effective for
financial statements issued subsequent to November 15,
2007. We do not expect that the adoption of this standard will
have a material impact on our consolidated financial position,
results of operations or cash flows.
In September 2006, the Securities and Exchange Commission
(“SEC”) issued Staff Accounting Bulletin 108,
Considering the Effects on Prior Year Misstatements when
Quantifying Misstatements in Current Year Financial Statements,
(“SAB 108”). SAB 108 requires registrants to
quantify errors using both the income statement method (i.e.
iron curtain method) and the rollover method and requires
adjustment if either method indicates a material error. If a
correction in the current year relating to prior year errors is
material to the current year, then the prior year financial
information needs to be corrected. A correction to the prior
year results that are not material to those years, would not
require a restatement process where prior financials would be
amended. SAB 108 is effective for fiscal years ending after
November 15, 2006. Our adoption of SAB 108 did not
have a material effect on our financial position, results of
operations or cash flows.
In December 2006, the FASB issued FSP
EITF 00-19-2
“Accounting for Registration Payment Arrangements”
(“FSP
EITF 00-19-2”).
FSP
EITF 00-19-2
addresses an issuer’s accounting for registration payment
arrangements.
58
This pronouncement specifies that the contingent obligation to
make future payments or otherwise transfer consideration under a
registration payment arrangement, whether issued as a separate
agreement or included as a provision of a financial instrument,
should be separately recognized and accounted for as a
contingency in accordance with SFAS 5 “Accounting for
Contingencies.” FSP
EITF 00-19-2
amending previous standards relating to rights agreements became
effective on December 21, 2006 with respect to arrangements
entered into or modified beginning on such date and for the
first fiscal year beginning after December 15, 2006 with
respect to those arrangements entered into prior to
December 21, 2006. We are in the process of evaluating the
impact of the adoption of this statement on our results of
operations and financial condition.
Other accounting standards that have been issued or proposed by
the FASB or other standards-setting bodies that do not require
adoption until a future date are not expected to have a material
impact on the consolidated financial statements upon adoption.
ITEM 7A. Quantitative
and Qualitative Disclosures About Market Risk
Interest
Rate Market Risk
Our exposure to interest rate risk is confined to our excess
cash in highly liquid money market funds denominated in
U.S. dollars. The primary objective of our investment
activities is to preserve our capital to fund operations. We
also seek to maximize income from our investments without
assuming significant risk. We do not use derivative financial
instruments in our investment portfolio. Our cash and
investments policy emphasizes liquidity and preservation of
principal over other portfolio considerations.
Foreign
Currency Market Risks
We have two significant subsidiaries, one each in Europe and
Mexico. Revenues and expenses associated with these subsidiaries
are denominated in foreign currency. Accordingly, our operating
results are affected by exchange rate fluctuations between the
U.S. dollar and these foreign currencies In order to
mitigate our exposure to foreign currency rate fluctuations, we
maintain minimal cash balances in the foreign subsidiaries.
However, if we are successful in our efforts to grow
internationally, our exposure to foreign currency rate
fluctuations, primarily the Euro and Mexican Peso, may increase.
We are also exposed to foreign currency risk related to the Euro
denominated and Mexican Peso denominated intercompany
receivables. Because our intercompany receivables are accounted
for in Euros and US dollars, any appreciation or devaluation of
the Euro or Mexican Peso will result in a gain or loss to the
consolidated statements of operations.
We do not currently enter into forward exchange contracts to
hedge exposure denominated in foreign currencies or any other
derivative financial instrument for trading or speculative
purposes. In the future, if we believe our currency exposure
merits, we may consider entering into transactions to help
mitigate the risk.
59
|
|
|
ITEM 8.
|
Consolidated
Financial Statements and Supplementary Data
|
Oculus
Innovative Sciences, Inc.
Index to
Consolidated Financial Statements
60
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board
of Directors and Stockholders of
Oculus Innovative Sciences, Inc. and Subsidiaries
We have audited the accompanying consolidated balance sheets of
Oculus Innovative Sciences, Inc. and Subsidiaries (the
“Company”) as of March 31, 2007 and 2006, and the
related consolidated statements of operations and comprehensive
loss and stockholders’ equity (deficit) and cash flows for
each of the three years in the period ended March 31, 2007.
These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these consolidated financial statements based on our
audit.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. The Company is not required to
have, nor were we engaged to perform, an audit of its internal
control over financial reporting. Our audit included
consideration of internal control over financial reporting as a
basis for designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred
to above present fairly, in all material respects, the
consolidated financial position of Oculus Innovative Sciences,
Inc. and Subsidiaries as of March 31, 2007 and 2006, and
the results of their operations and their cash flows for each of
the three years in the period ended March 31, 2007 in
conformity with United States generally accepted accounting
principles.
/s/
Marcum & Kliegman LLP
New York, New York
June 13, 2007
61
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED
BALANCE SHEETS
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
(In thousands, except per share amounts)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
19,050
|
|
|
$
|
7,448
|
|
|
Restricted cash
|
|
|
2,000
|
|
|
|
—
|
|
|
Accounts receivable, net
|
|
|
1,364
|
|
|
|
1,076
|
|
|
Inventories
|
|
|
282
|
|
|
|
317
|
|
|
Prepaid expenses and other current
assets
|
|
|
1,172
|
|
|
|
1,386
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
23,868
|
|
|
|
10,227
|
|
|
Property and equipment, net
|
|
|
2,207
|
|
|
|
1,940
|
|
|
Restricted cash
|
|
|
49
|
|
|
|
44
|
|
|
Deferred offering costs
|
|
|
—
|
|
|
|
478
|
|
|
Debt issue costs, net
|
|
|
826
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
26,950
|
|
|
$
|
12,689
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND
STOCKHOLDERS’
EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
2,551
|
|
|
$
|
2,774
|
|
|
Accrued expenses and other current
liabilities
|
|
|
1,421
|
|
|
|
1,686
|
|
|
Dividends payable
|
|
|
—
|
|
|
|
121
|
|
|
Current portion of long-term debt
|
|
|
6,045
|
|
|
|
504
|
|
|
Current portion of capital lease
obligations
|
|
|
17
|
|
|
|
15
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
10,034
|
|
|
|
5,100
|
|
|
Long-term debt, less current
portion
|
|
|
1,990
|
|
|
|
210
|
|
|
Capital lease obligations, less
current portion
|
|
|
25
|
|
|
|
41
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
12,049
|
|
|
|
5,351
|
|
|
|
|
|
|
|
|
|
|
|
|
Commitments and Contingencies
|
|
|
|
|
|
|
|
|
|
Stockholders’ Equity
|
|
|
|
|
|
|
|
|
|
Convertible preferred stock,
$0.0001 par value; 30,000,000 shares authorized,
Series A, none and 1,347,709 shares issued and
outstanding at March 31, 2007 and 2006, respectively
|
|
|
—
|
|
|
|
6,668
|
|
|
Series B, none and
2,635,744 shares issued and outstanding at March 31,
2007 and 2006, respectively
|
|
|
—
|
|
|
|
43,722
|
|
|
Common stock, $0.0001 par
value; 100,000,000 shares authorized,
|
|
|
—
|
|
|
|
|
|
|
11,844,411 and
4,218,981 shares issued and outstanding at March 31,
2007 and
|
|
|
|
|
|
|
|
|
|
2006, respectively
|
|
|
1
|
|
|
|
3,399
|
|
|
Additional paid-in capital
|
|
|
85,751
|
|
|
|
4,644
|
|
|
Deferred compensation
|
|
|
—
|
|
|
|
(798
|
)
|
|
Accumulated other comprehensive
income (loss)
|
|
|
(364
|
)
|
|
|
3
|
|
|
Accumulated deficit
|
|
|
(70,487
|
)
|
|
|
(50,300
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
14,901
|
|
|
|
7,338
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and
stockholders’ equity
|
|
$
|
26,950
|
|
|
$
|
12,689
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying footnotes are an integral part of these
financial statements.
62
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands, except
|
|
|
|
|
per share amounts)
|
|
|
|
|
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product
|
|
$
|
3,679
|
|
|
$
|
1,966
|
|
|
$
|
473
|
|
|
Service
|
|
|
864
|
|
|
|
618
|
|
|
|
883
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
4,543
|
|
|
|
2,584
|
|
|
|
1,356
|
|
|
Cost of revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product
|
|
|
2,104
|
|
|
|
3,899
|
|
|
|
2,211
|
|
|
Service
|
|
|
895
|
|
|
|
1,003
|
|
|
|
1,311
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total cost of revenues
|
|
|
2,999
|
|
|
|
4,902
|
|
|
|
3,522
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit (loss)
|
|
|
1,544
|
|
|
|
(2,318
|
)
|
|
|
(2,166
|
)
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
4,508
|
|
|
|
2,600
|
|
|
|
1,654
|
|
|
Selling, general and administrative
|
|
|
16,520
|
|
|
|
15,933
|
|
|
|
12,492
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
21,028
|
|
|
|
18,533
|
|
|
|
14,146
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations
|
|
|
(19,484
|
)
|
|
|
(20,851
|
)
|
|
|
(16,312
|
)
|
|
Interest expense
|
|
|
(956
|
)
|
|
|
(172
|
)
|
|
|
(372
|
)
|
|
Interest income
|
|
|
312
|
|
|
|
282
|
|
|
|
8
|
|
|
Other income (expense), net
|
|
|
345
|
|
|
|
(377
|
)
|
|
|
146
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss from continuing operations
|
|
|
(19,783
|
)
|
|
|
(21,118
|
)
|
|
|
(16,530
|
)
|
|
Loss from operations of
discontinued business
|
|
|
—
|
|
|
|
(818
|
)
|
|
|
—
|
|
|
Loss on disposal of discontinued
business
|
|
|
—
|
|
|
|
(1,163
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss on discontinued operations
|
|
|
—
|
|
|
|
(1,981
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(19,783
|
)
|
|
|
(23,099
|
)
|
|
|
(16,530
|
)
|
|
Preferred stock dividends
|
|
|
(404
|
)
|
|
|
(121
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss available to common
stockholders
|
|
$
|
(20,187
|
)
|
|
$
|
(23,220
|
)
|
|
$
|
(16,530
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share: basic
and diluted
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Continuing operations
|
|
$
|
(3.71
|
)
|
|
$
|
(5.12
|
)
|
|
$
|
(4.22
|
)
|
|
Discontinued operations
|
|
|
—
|
|
|
|
(0.48
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
(3.71
|
)
|
|
$
|
(5.60
|
)
|
|
$
|
(4.22
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-average number of shares
used in per common share calculations:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted
|
|
|
5,448
|
|
|
|
4,150
|
|
|
|
3,914
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive loss, net of
tax
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(19,783
|
)
|
|
$
|
(23,099
|
)
|
|
$
|
(16,530
|
)
|
|
Foreign currency translation
adjustments
|
|
|
(367
|
)
|
|
|
144
|
|
|
|
(127
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
$
|
(20,150
|
)
|
|
$
|
(22,955
|
)
|
|
$
|
(16,657
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying footnotes are an integral part of these
financial statements.
63
OCULUS
INNOVATIVE SCIENCES, INC.
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumu-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred
|
|
|
lated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Addi-
|
|
|
Stock-
|
|
|
Other
|
|
|
|
|
|
|
|
|
|
|
Convertible Preferred Stock
|
|
|
|
|
|
|
|
|
tional
|
|
|
Based
|
|
|
Comprehensive
|
|
|
Accum-
|
|
|
|
|
|
|
|
Series A ($0.0001 par Value)
|
|
|
Series B ($0.0001 par Value)
|
|
|
Series C ($0.0001 par Value)
|
|
|
Common Stock ($0.0001 par Value)
|
|
|
Paid in
|
|
|
Compen-
|
|
|
Income
|
|
|
ulated
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
sation
|
|
|
(Loss)
|
|
|
Deficit
|
|
|
Total
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Balance, March 31, 2004
|
|
|
1,337,709
|
|
|
$
|
6,628
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,914,653
|
|
|
$
|
3,101
|
|
|
$
|
661
|
|
|
$
|
(208
|
)
|
|
$
|
(14
|
)
|
|
$
|
(10,550
|
)
|
|
$
|
(382
|
)
|
|
Deferred stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,765
|
|
|
|
(2,765
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Amortization of stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,297
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,297
|
|
|
Non-employee stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
30
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
30
|
|
|
Reclassification of options subject
to cash settlement
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
113
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
113
|
|
|
Issuance of common stock warrants
in connection with debt financing
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
28
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
28
|
|
|
Issuance of Series A
convertible preferred stock warrants in connection with debt
financing
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
77
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
77
|
|
|
Issuance of Series B
convertible preferred stock, net of offering costs
|
|
|
—
|
|
|
|
—
|
|
|
|
1,014,093
|
|
|
|
16,696
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
16,696
|
|
|
Foreign currency translation
adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(127
|
)
|
|
|
—
|
|
|
|
(127
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(16,530
|
)
|
|
|
(16,530
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, March 31, 2005
|
|
|
1,337,709
|
|
|
|
6,628
|
|
|
|
1,014,093
|
|
|
|
16,696
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,914,653
|
|
|
|
3,101
|
|
|
|
3,674
|
|
|
|
(676
|
)
|
|
|
(141
|
)
|
|
|
(27,080
|
)
|
|
|
2,202
|
|
|
Issuance of common stock upon
exercise of stock options
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
291,828
|
|
|
|
298
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
298
|
|
|
Deferred stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
401
|
|
|
|
(401
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Amortization of stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
279
|
|
|
|
—
|
|
|
|
—
|
|
|
|
279
|
|
|
Non-employee stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
32
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
32
|
|
|
Fair value related to common stock
warrants with service conditions
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
153
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
153
|
|
|
Issuance of common stock in
exchange for services
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,500
|
|
|
|
—
|
|
|
|
127
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
127
|
|
|
Reclassification of options subject
to cash settlement
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
257
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
257
|
|
|
Issuance of Series B
convertible preferred stock, net of offering costs
|
|
|
—
|
|
|
|
—
|
|
|
|
1,621,651
|
|
|
|
27,026
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
27,026
|
|
|
Issuance of Series A
convertible preferred stock in connections with convertible debt
|
|
|
10,000
|
|
|
|
40
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
40
|
|
|
Dividend payable to Series A
convertible preferred stockholders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(121
|
)
|
|
|
(121
|
)
|
|
Foreign currency translation
adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
144
|
|
|
|
—
|
|
|
|
144
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(23,099
|
)
|
|
|
(23,099
|
)
|
|
Balance, March 31, 2006
|
|
|
1,347,709
|
|
|
$
|
6,668
|
|
|
|
2,635,744
|
|
|
$
|
43,722
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,218,981
|
|
|
$
|
3,399
|
|
|
$
|
4,644
|
|
|
$
|
(798
|
)
|
|
$
|
3
|
|
|
$
|
(50,300
|
)
|
|
$
|
7,338
|
|
64
OCULUS
INNOVATIVE SCIENCES, INC.
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumu-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred
|
|
|
lated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Addi-
|
|
|
Stock-
|
|
|
Other
|
|
|
|
|
|
|
|
|
|
|
Convertible Preferred Stock
|
|
|
|
|
|
|
|
|
tional
|
|
|
Based
|
|
|
Comprehensive
|
|
|
Accum-
|
|
|
|
|
|
|
|
Series A ($0.0001 par Value)
|
|
|
Series B ($0.0001 par Value)
|
|
|
Series C ($0.0001 par Value)
|
|
|
Common Stock ($0.0001 par Value)
|
|
|
Paid in
|
|
|
Compen-
|
|
|
Income
|
|
|
ulated
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
sation
|
|
|
(Loss)
|
|
|
Deficit
|
|
|
Total
|
|
|
|
|
(In thousands, except share and per share amounts)
|
|
|
|
|
Issuance of common stock in
connection with IPO and exercise of over-allotment, net of
discounts, commissions, expenses and other offering costs
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,353,550
|
|
|
|
|
|
|
|
21,936
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
21,936
|
|
|
Issuance of common stock in
connection with exercise of warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,138
|
|
|
|
—
|
|
|
|
21
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
21
|
|
|
Issuance of common stock in
connection with services rendered
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,750
|
|
|
|
—
|
|
|
|
43
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
43
|
|
|
Issuance of Series C
convertible preferred stock, net of offering costs
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
193,045
|
|
|
|
2,903
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,903
|
|
|
Conversion of convertible preferred
stock into common stock at the closing of the IPO on
January 30, 2007
|
|
|
(1,347,709
|
)
|
|
|
(6,668
|
)
|
|
|
(2,635,744
|
)
|
|
|
(43,722
|
)
|
|
|
(193,045
|
)
|
|
|
(2,903
|
)
|
|
|
4,176,498
|
|
|
|
—
|
|
|
|
53,293
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Reclassification to APIC in
connection with Delaware reincorporation due to change in par
value
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,398
|
)
|
|
|
3,398
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Reclassification of deferred
stock-based compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(798
|
)
|
|
|
798
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Amortization of stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
158
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
158
|
|
|
Non-employee stock-based
compensation
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
11
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
11
|
|
|
Employee stock-based compensation
expense recognized under SFAS No. 123R, net of
forfeitures
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
815
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
815
|
|
|
Fair value adjustment related to
common stock warrants with service conditions
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
555
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
555
|
|
|
Issuance of common warrants in
connection with debt financing
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,150
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,150
|
|
|
Dividend payable to Series A
convertible preferred stockholders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(404
|
)
|
|
|
(404
|
)
|
|
Common stock dividend paid to
Series A convertible preferred stockholders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
87,494
|
|
|
|
—
|
|
|
|
525
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
525
|
|
|
Foreign currency translation
adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
—
|
|
|
|
(367
|
)
|
|
|
|
|
|
|
(367
|
)
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(19,783
|
)
|
|
|
(19,783
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, March 31, 2007
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
11,844,411
|
|
|
$
|
1
|
|
|
$
|
85,751
|
|
|
|
—
|
|
|
$
|
(364
|
)
|
|
$
|
(70,487
|
)
|
|
$
|
14,901
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying footnotes are an integral part of these
financial statements.
65
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
CONSOLIDATED
STATEMENTS OF CASH FLOWS
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash flows from operating
activities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss from continuing operations
|
|
$
|
(19,783
|
)
|
|
$
|
(21,118
|
)
|
|
$
|
(16,530
|
)
|
|
Adjustments to reconcile net loss
to net cash used in operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization
|
|
|
672
|
|
|
|
651
|
|
|
|
434
|
|
|
Stock-based compensation
|
|
|
1,582
|
|
|
|
597
|
|
|
|
2,349
|
|
|
Non-cash interest expense
|
|
|
547
|
|
|
|
21
|
|
|
|
131
|
|
|
Unrealized foreign exchange gain
|
|
|
(407
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Loss on disposal of assets
|
|
|
—
|
|
|
|
113
|
|
|
|
2
|
|
|
Changes in operating assets and
liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable
|
|
|
(287
|
)
|
|
|
(849
|
)
|
|
|
217
|
|
|
Inventories
|
|
|
53
|
|
|
|
551
|
|
|
|
(748
|
)
|
|
Prepaid expenses and other current
assets
|
|
|
219
|
|
|
|
(887
|
)
|
|
|
(278
|
)
|
|
Accounts payable
|
|
|
(245
|
)
|
|
|
1,868
|
|
|
|
(165
|
)
|
|
Accrued expenses and other
liabilities
|
|
|
(433
|
)
|
|
|
(649
|
)
|
|
|
1,055
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating
activities
|
|
|
(18,082
|
)
|
|
|
(19,702
|
)
|
|
|
(13,533
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment
|
|
|
(873
|
)
|
|
|
(475
|
)
|
|
|
(1,042
|
)
|
|
Issuance of note receivable
|
|
|
—
|
|
|
|
55
|
|
|
|
(55
|
)
|
|
Changes in restricted cash
|
|
|
(4
|
)
|
|
|
1
|
|
|
|
(21
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing
activities
|
|
|
(877
|
)
|
|
|
(419
|
)
|
|
|
(1,118
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred offering costs
|
|
|
478
|
|
|
|
(478
|
)
|
|
|
—
|
|
|
Proceeds from issuance of common
stock, net of offering costs
|
|
|
21,936
|
|
|
|
—
|
|
|
|
—
|
|
|
Proceeds from issuance of common
stock upon exercise of stock options and warrants
|
|
|
21
|
|
|
|
298
|
|
|
|
—
|
|
|
Proceeds from issuance of
convertible preferred stock
|
|
|
2,903
|
|
|
|
27,026
|
|
|
|
16,696
|
|
|
Debt issuance costs
|
|
|
(77
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Cash restricted for repayment of
debt
|
|
|
(2,000
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Proceeds from issuance of debt
|
|
|
9,056
|
|
|
|
257
|
|
|
|
1,205
|
|
|
Principal payments on debt
|
|
|
(1,734
|
)
|
|
|
(953
|
)
|
|
|
(664
|
)
|
|
Payments on capital lease
obligations
|
|
|
(15
|
)
|
|
|
(31
|
)
|
|
|
(41
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing
activities
|
|
|
30,568
|
|
|
|
26,119
|
|
|
|
17,196
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from discontinued
operations
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating cash flows
|
|
|
—
|
|
|
|
(818
|
)
|
|
|
—
|
|
|
Investing cash flows
|
|
|
—
|
|
|
|
(1,163
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in discontinued
operations
|
|
|
—
|
|
|
|
(1,981
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effect of exchange rate on cash
and cash equivalents
|
|
|
(7
|
)
|
|
|
144
|
|
|
|
(127
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase in cash and cash
equivalents
|
|
|
11,602
|
|
|
|
4,161
|
|
|
|
2,418
|
|
|
Cash and cash equivalents,
beginning of year
|
|
|
7,448
|
|
|
|
3,287
|
|
|
|
869
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents, end of
year
|
|
$
|
19,050
|
|
|
$
|
7,448
|
|
|
$
|
3,287
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash
flow information:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
391
|
|
|
$
|
125
|
|
|
$
|
221
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash investing and
financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equipment acquired under capital
leases
|
|
|
—
|
|
|
|
—
|
|
|
$
|
37
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of note payable into
Series A convertible preferred stock
|
|
|
—
|
|
|
$
|
40
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair value of warrants issued in
connection with debt
|
|
$
|
1,150
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying footnotes are an integral part of these
financial statements.
66
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
Organization
Oculus Innovative Sciences, Inc. (the “Company”) was
incorporated under the laws of the State of California in April
1999 and was reincorporated under the laws of the State of
Delaware in December 2006. The Company’s principal office
is located in Petaluma, California. The Company develops,
manufactures and markets a family of products intended to
prevent and treat infections in chronic and acute wounds. The
Company’s platform technology, called Microcyn, is a
proprietary oxychlorine small molecule formulation that is
designed to treat a wide range of organisms that cause disease,
or pathogens, including viruses, fungi, spores and antibiotic
resistant strains of bacteria. The Company conducts its business
worldwide, with significant operating subsidiaries in Europe and
Mexico.
Delaware
Reincorporation
On December 15, 2006, the Company merged into OIS
Reincorporation Sub, Inc., a Delaware corporation (the Delaware
Company). Pursuant to the Merger Agreement, an amendment to the
certificate of incorporation was filed pursuant to which
(i) each four shares of outstanding Company common stock
were converted into one share of the Delaware Company’s
common stock ($0.0001 par value), (ii) each four
shares of the Company’s outstanding Series A
convertible preferred stock were converted into one share of the
Delaware Company’s Series A convertible preferred
stock ($0.0001 par value), (iii) each four shares of
the Company’s outstanding Series B convertible
preferred stock were converted into one share of the Delaware
Company’s Series B convertible preferred stock
($0.0001 par value), and (iv) each four shares of the
California Company’s outstanding Series C convertible
preferred stock were converted into one share of the Delaware
Company’s Series C convertible preferred stock
($0.0001 par value). In addition, all options, warrants or
rights to purchase shares of Company common stock or Company
convertible preferred stock outstanding immediately prior to the
Reincorporation were converted into options, warrants or rights
to purchase an equivalent number of shares of the Delaware
Company’s common stock or convertible preferred stock, as
the case may be, and those securities will continue to vest upon
the same terms and conditions as existed immediately prior to
the Reincorporation.
Incorporation
of Oculus Japan
In October 2006, the Company incorporated Oculus Innovative
Sciences K.K., (“Oculus Japan”) a wholly owned
subsidiary. The Japanese subsidiary will primarily conduct
research and development activities. Oculus Japan is
insignificant with respect to the Company’s consolidated
operating results for the year ended March 31, 2007.
Reverse
Stock Split
On December 15, 2006, the Company effected a
1-for-4
reverse split of its common stock and convertible preferred
stock. All common and convertible preferred shares and per share
amounts have been retroactively restated in the accompanying
consolidated financial statements and notes for all periods
presented.
|
|
|
NOTE 2 —
|
Liquidity
and Financial Condition
|
The Company incurred net losses of $19,783,000, $23,099,000, and
$16,530,000 for the years ended March 31, 2007, 2006 and
2005, respectively. At March 31, 2007, the Company’s
accumulated deficit amounted to $70,489,000.
Through March 31, 2007, the Company raised, net of offering
costs, an aggregate of approximately $85,752,000 in various
equity financing transactions that, together with the proceeds
of certain debt financing transactions, enabled it to sustain
operations while attempting to execute its business plan. The
Company had working capital of $13,834,000 as of March 31,
2007.
67
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
In June 2006, the Company entered into a $5,000,000 credit
facility from which it drew $4,182,000, to fund its operations,
and invest in new equipment. In addition, on November 7,
2006, the Company entered into a $4,000,000 Bridge Loan
agreement which becomes due on November 7, 2007 with
Mr. Robert Burlingame, one of the Company’s directors
(Note 10).
The Company’s Board of Directors and stockholders approved
an amendment to the Articles of Incorporation (that became
effective on August 28, 2006) to authorize the
issuance of up to 875,000 shares of Series C
convertible preferred stock. On September 14, 2006 and
October 20, 2006 the Company sold an aggregate of
193,045 units, consisting of 193,045 shares of
Series C convertible preferred stock and warrants to
purchase 38,604 shares of the Company’s common stock.
Net proceeds from the offering, after deducting commissions were
$2,903,000 (Note 13).
The Company’s Registration Statement on
Form S-1,
Amendment No. 7, (File
No. 333-135584)
related to the Company’s initial public offering was
declared effective by the SEC on January 24, 2007. A total
of 3,025,000 shares of the Company’s common stock were
registered with the SEC. All of these shares were registered on
the Company’s behalf. The offering commenced on
January 25, 2007 and 3,025,000 shares of common stock
offered were sold on January 30, 2007 for an aggregate
offering price of $24,200,000 through the managing underwriters:
Roth Capital Partners, Maxim LLC and Brookstreet Securities
Corporation.
On February 16, 2007 the underwriters of the Company’s
initial public offering exercised a portion of their
over-allotment option and purchased 328,550 shares of the
Company’s common stock in accordance with the terms of the
underwriting agreement for an aggregate offering price of
$2,628,000 through the managing underwriters: Roth Capital
Partners, Maxim LLC and Brookstreet Securities Corporation.
The Company paid to the underwriters underwriting discounts,
commissions and non-accountable expenses totaling $2,146,000 in
connection with the initial public offering and the underwriters
exercise of the over-allotment shares. In addition, the Company
incurred additional expenses of approximately $2,746,000 in
connection with the initial public offering, which when added to
the underwriting discounts, commissions and non-accountable
expenses paid by the Company amounts to total expenses of
$4,892,000. Thus the net offering proceeds to the Company (after
deducting underwriting discounts and commissions and offering
expenses) were approximately $21,936,000.
The Company currently intends to use the proceeds of the initial
public offering to fund its sales and marketing activities,
clinical trials and related research. The remaining proceeds are
to be used for general corporate purposes, including working
capital. The Company anticipates it will incur significant costs
in connection with Sarbanes-Oxley compliance and other costs
associated with reporting as a public entity.
The Company currently anticipates that its cash, cash
equivalents, and restricted cash balances, together with
revenues it expects to generate and interest it expects to earn
on invested funds will be sufficient to meet its anticipated
cash requirements through at least April 1, 2008. The
Company also expects to continue incurring losses for the
foreseeable future and must raise substantial additional capital
during the year ending March 31, 2008 to pursue its product
development initiatives, fund clinical trials and penetrate
markets for the sale of its products. The Company is currently
planning to commence Phase 3 clinical trials of its Microcyn
products. Management considers the execution and eventual
completion of these trials to be a critical milestone in the
development of the business. These clinical trials are likely to
be lengthy and expensive and cannot be commenced during the year
ending March 31, 2008 unless the Company raises additional
capital. These clinical trials must also be completed in order
for the Company to commercialize Microcyn as a drug product in
the United States.
Management believes that the Company has access to capital
resources through possible public or private equity offerings,
debt financings, corporate collaborations or other means;
however, the Company has not secured any commitment for new
financing at this time nor can it provide any assurance that new
financing will be available on commercially acceptable terms, if
at all. If the Company is unable to secure additional capital,
it will be required to curtail it’s research and
development initiatives, delay clinical trials and take
additional measures to reduce costs
68
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
in order to conserve its cash. These measures could cause
significant delays in the Company’s efforts to
commercialize its products in the United States, which is
critical to the realization of its business plan and the future
operations of the Company.
|
|
|
NOTE 3 —
|
Summary
of Significant Accounting Policies
|
Principles
of Consolidation
The accompanying consolidated financial statements include the
accounts of the Company and its wholly-owned subsidiaries,
Aquamed Technologies, Inc., Oculus Technologies of Mexico C.V.
(“OTM”), Oculus Innovative Sciences B.V. (“OIS
Europe”), and Oculus Innovative Sciences K.K. (“OIS
Japan”). All significant intercompany accounts and
transactions have been eliminated in consolidation.
The consolidated financial statements are presented in United
States Dollars in accordance with Statement of Financial
Accounting Standard (“SFAS”) No. 52,
“Foreign Currency Translation”
(“SFAS 52”). The Company’s subsidiary OTM
uses the local currency (Mexican Pesos) as its functional
currency, OIS Europe uses the local currency (Euro) as its
functional currency and OIS Japan uses the local currency (Yen)
as its functional currency. Assets and liabilities are
translated at exchange rates in effect at the balance sheet date
and revenue and expense accounts are translated at average
exchange rates during the period. Resulting translation
adjustments are recorded directly to accumulated other
comprehensive (loss) income. Loans made to subsidiaries OTM and
OIS Europe will be paid back to the Company in the future when
subsidiaries begin to generate cash.
The Company, in determining whether it is required to
consolidate investee businesses, considers both the voting and
variable interest models of consolidation as required under
Financial Accounting Standards Board (“FASB”)
Interpretation No. 46(R) “Consolidation of Variable
Interest Entities,” (“FIN 46(R)”).
Accordingly the Company consolidates investee entities when it
owns less than 50% of the voting interests but, based on the
risks and rewards of its participation has established financial
control. As described in Note 18, the Company’s
consolidated financial statements for the year ended
March 31, 2006 include the results of a variable interest
entity that is being presented as a discontinued operation in
accordance with SFAS No. 144 “Accounting for the
Impairment and Disposal of Long Lived Assets,”
(“SFAS 144”).
Use of
Estimates
The preparation of consolidated financial statements in
conformity with accounting principles generally accepted in the
United States of America requires management to make estimates
and assumptions that affect the reported amounts of assets and
liabilities and disclosures of contingent liabilities at the
dates of the consolidated financial statements and the reported
amounts of revenues and expenses during the reporting periods.
Actual results could differ from these estimates. Significant
estimates and assumptions include reserves and write-downs
related to receivables and inventories, the recoverability of
long-term assets, deferred taxes and related valuation
allowances and valuation of equity instruments.
Revenue
Recognition
The Company generates revenue from sales of its products to
hospitals, medical centers, doctors, pharmacies, and
distributors. The Company sells its products directly to third
parties and to distributors through various cancelable
distribution agreements. The Company has also entered into
agreements to license its technology.
The Company also provides regulatory compliance testing and
quality assurance services to medical device and pharmaceutical
companies.
The Company applies the revenue recognition principles set forth
in Securities and Exchange Commission Staff Accounting Bulletin
(“SAB”) 104 “Revenue Recognition” with
respect to all of its revenue. Accordingly, the
69
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Company records revenue when (i) persuasive evidence of an
arrangement exists, (ii) delivery has occurred,
(iii) the fee is fixed or determinable, and
(iv) collectability of the sale is reasonably assured.
The Company requires all of its product sales to be supported by
evidence of a sale transaction that clearly indicates the
selling price to the customer, shipping terms and payment terms.
Evidence of an arrangement generally consists of a contract or
purchase order approved by the customer. The Company has ongoing
relationships with certain customers from which it customarily
accepts orders by telephone in lieu of a purchase order.
The Company recognizes revenue at the time in which it receives
a confirmation that the goods were either tendered at their
destination when shipped “FOB destination,” or
transferred to a shipping agent when shipped “FOB shipping
point.” Delivery to the customer is deemed to have occurred
when the customer takes title to the product. Generally, title
passes to the customer upon shipment, but could occur when the
customer receives the product based on the terms of the
agreement with the customer.
The selling prices of all goods that the Company sells are
fixed, and agreed to with the customer, prior to shipment.
Selling prices are generally based on established list prices.
The Company does not customarily permit its customers to return
any of its products for monetary refunds or credit against
completed or future sales. The Company, from time to time, may
replace expired goods on a discretionary basis. The Company
records these types of adjustments, when made, as a reduction of
revenue. Sales adjustments were insignificant during the years
ended March 31, 2007, 2006 and 2005.
The Company evaluates the creditworthiness of new customers and
monitors the creditworthiness of its existing customers to
determine whether events or changes in their financial
circumstances would raise doubt as to the collectability of a
sale at the time in which a sale is made. Payment terms on sales
made in the United States are generally 30 days and
internationally, generally range from 30 days to
180 days.
In the event a sale is made to a customer under circumstances in
which collectability is not reasonably assured, the Company
either requires the customer to remit payment prior to shipment
or defers recognition of the revenue until the time of
collection. The Company maintains a reserve for amounts which
may not be collectible due to risk of credit losses.
During the fiscal year ended March 31, 2005, approximately
$434,000 of sales in Mexico was recognized when cash was
collected since collection was not reasonably assured.
The Company has entered into distribution agreements in Europe.
Recognition of revenue and related cost of revenue from product
sales is deferred until the product is sold from the
distributors to their customers.
When the Company receives letters of credit and the terms of the
sale provide for no right of return except to replace defective
product, revenue is recognized when the letter of credit becomes
effective and the product is shipped.
Revenue from consulting contracts is recognized as services are
provided. Revenue from testing contracts is recognized as tests
are completed and a final report is sent to the customer.
Cash
and Cash Equivalents
The Company considers all highly liquid investments with an
original maturity of three months or less when purchased to be
cash equivalents. Cash equivalents may be invested in money
market funds, commercial paper, variable rate demand
instruments, and certificates of deposits. Cash equivalents are
carried at cost, which approximates fair value.
70
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Restricted
Cash
On March 29, 2007, the Company entered into Amendment
No. 1 to the Bridge Loan with Mr. Robert Burlingame,
one of the Company’s directors. Pursuant to the Amendment,
the Company deposited $2,000,000 into a segregated
interest-bearing account and is reported in current assets in
the accompanying consolidated balance sheet (Note 10).
In connection with certain operating lease agreements
(Note 12), the Company is required to maintain cash
deposits in a restricted account. Restricted cash held as
security under this arrangement amounted to $49,000 and $44,000
at March 31, 2007 and 2006, respectively and is reported in
non-current assets in the accompanying consolidated balance
sheets as restricted cash.
Concentration
of Credit Risk
Financial instruments that potentially subject the Company to
concentration of credit risk consist principally of cash, cash
equivalents and accounts receivable. Cash and cash equivalents
are maintained in financial institutions in the United States,
Mexico, The Netherlands and Japan. The Company is exposed to
credit risk in the event of default by these financial
institutions for amounts in excess of the Federal Deposit
Insurance Corporation insured limits. Management believes that
the financial institutions that hold the Company’s deposits
are financially sound and have minimal credit risk. Cash and
cash equivalents held in foreign banks are intentionally kept at
minimal levels, and therefore have minimal credit risk
associated with them.
The Company grants credit to its business customers, which are
primarily located in Mexico, Europe and the United States.
Collateral is generally not required for trade receivables. The
Company maintains allowances for potential credit losses. One
customer represented 12% of the net accounts receivable balance
at March 31, 2007. In the years ended March 31, 2007
and March 31, 2005, one customer in each year represented
13% and 15% of sales in each of the years, respectively.
Accounts
Receivable
Trade accounts receivable are recorded net of allowances for
cash discounts for prompt payment, doubtful accounts, and sales
returns. Estimates for cash discounts, and sales returns are
based on contractual terms, historical trends and expectations
regarding the utilization rates for these programs. With respect
to government chargebacks, the Mexican Ministry of Health’s
(“MOH”) policy is to levy penalties on its vendors for
product received after scheduled delivery times. The Company has
not incurred any such chargebacks to date; however such
penalties (if incurred) would be recorded as a reduction of
revenue and the related accounts receivable balance.
The Company’s policy is to reserve for uncollectible
accounts based on its best estimate of the amount of probable
credit losses in its existing accounts receivable. The Company
periodically reviews its accounts receivable to determine
whether an allowance for doubtful accounts is necessary based on
an analysis of past due accounts and other factors that may
indicate that the realization of an account may be in doubt.
Other factors that the Company considers include its existing
contractual obligations, historical payment patterns of its
customers and individual customer circumstances, an analysis of
days sales outstanding by customer and geographic region, and a
review of the local economic environment and its potential
impact on government funding and reimbursement practices.
Account balances deemed to be uncollectible are charged to the
allowance after all means of collection have been exhausted and
the potential for recovery is considered remote. The Company had
a low occurrence of credit losses through the year ended
March 31, 2005 and therefore did not consider it necessary
to establish an allowance for doubtful accounts as of
March 31, 2005. The allowance for doubtful accounts at
March 31, 2007 and 2006 represents probable credit losses
in the amounts of $207,000 and $90,000, respectively.
71
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Inventories
Inventories are stated at the lower of cost, cost being
determined on a standard cost basis (which approximates actual
cost on a
first-in,
first-out basis), or market.
Due to changing market conditions, estimated future
requirements, age of the inventories on hand and production of
new products, the Company regularly reviews inventory quantities
on hand and records a provision to write down excess and
obsolete inventory to its estimated net realizable value. The
Company recorded reserves to reduce the carrying amounts of
inventories to their net realizable value in the amounts of
$94,000 and $996,000 for the years ended March 31, 2007 and
2006, respectively. During the year ended March 31, 2007,
the Company disposed of inventory reserved in prior periods.
Property
and Equipment
Property and equipment are stated at cost less accumulated
depreciation and amortization. Depreciation of property and
equipment is computed using the straight-line method over the
estimated useful lives of the respective assets. Depreciation of
leasehold improvements is computed using the straight-line
method over the lesser of the estimated useful life of the
improvement or the remaining term of the lease. Estimated useful
asset life by classification is as follows:
| |
|
|
|
|
|
|
|
Years
|
|
|
|
|
Office equipment
|
|
|
3
|
|
|
Manufacturing and lab equipment
|
|
|
5
|
|
|
Furniture and fixtures
|
|
|
7
|
|
Upon retirement or sale, the cost and related accumulated
depreciation are removed from the consolidated balance sheet and
the resulting gain or loss is reflected in operations.
Maintenance and repairs are charged to operations as incurred.
Impairment
of Long-Lived Assets
The Company periodically reviews the carrying values of its long
lived assets in accordance with SFAS 144 “Long Lived
Assets” when events or changes in circumstances would
indicate that it is more likely than not that their carrying
values may exceed their realizable values, and records
impairment charges when considered necessary. Specific potential
indicators of impairment include, but are not necessarily
limited to:
a significant decrease in the fair value of an asset;
a significant change in the extent or manner in which an asset
is used or a significant physical change in an asset;
a significant adverse change in legal factors or in the business
climate that affects the value of an asset;
an adverse action or assessment by the U.S. Food and Drug
Administration or another regulator;
an accumulation of costs significantly in excess of the amount
originally expected to acquire or construct an asset; and
operating or cash flow losses combined with a history of
operating or cash flow losses or a projection or forecast that
demonstrates continuing losses associated with an
income-producing asset.
When circumstances indicate that an impairment may have
occurred, the Company tests such assets for recoverability by
comparing the estimated undiscounted future cash flows expected
to result from the use of such assets and their eventual
disposition to their carrying amounts. In estimating these
future cash flows, assets and liabilities are grouped at the
lowest level for which there are identifiable cash flows that
are largely independent of the cash flows generated by other
such groups. If the undiscounted future cash flows are less than
the carrying amount of the asset, an impairment loss, measured
as the excess of the carrying value of the asset over its
estimated
72
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
fair value, will be recognized. The cash flow estimates used in
such calculations are based on estimates and assumptions, using
all available information that management believes is reasonable.
Research
and Development
Research and development expense is charged to operations as
incurred and consists primarily of personnel expenses, clinical
and regulatory services and supplies. For the years ended
March 31, 2007, 2006 and 2005, research and development
expense amounted to $4,508,000, $2,600,000 and $1,654,000
respectively.
Advertising
Costs
Advertising costs are expensed as incurred. Advertising costs
amounted to $54,000, $126,000 and $122,000, for the years ended
March 31, 2007, 2006 and 2005, respectively. Advertising
costs are included in selling, general and administrative
expenses in the accompanying consolidated statements of
operations.
Shipping
and Handling Costs
The Company applies the guidelines enumerated in Emerging Issues
Task Force Issue (“EITF”)
00-10
“Accounting for Shipping and Handling Fees and Costs”
with respect to its shipping and handling costs. Accordingly,
the Company classifies amounts billed to customers related to
shipping and handling in sale transactions as revenue. Shipping
and handling costs incurred are recorded in cost of sales. To
date, shipping and handling costs billed to customers have been
insignificant.
Foreign
Currency Transactions
Foreign currency gains (losses) relate to working capital loans
that the Company has made to its foreign subsidiaries. The
Company recorded foreign currency gains (losses) for the years
ended March 31, 2007, 2006 and 2005 of $407,000, ($283,000)
and $134,000, respectively. The related gains (losses) were
recorded in other income (expense) in the accompanying
consolidated statements of operations.
Stock-Based
Compensation
Prior to April 1, 2006, the Company accounted for
stock-based employee compensation arrangements in accordance
with the provisions of APB No. 25, “Accounting for
Stock Issued to Employees,” and its related interpretations
and applied the disclosure requirements of
SFAS No. 148, “Accounting for Stock-Based
Compensation-Transition and Disclosure, an amendment of FASB
Statement No. 123.” The Company used the minimum value
method to measure the fair value of awards issued prior to
April 1, 2006 with respect to its application of the
disclosure requirements under SFAS No. 123.
Effective April 1, 2006, the Company adopted
SFAS No. 123(R) “Share Based Payment”
(“SFAS 123(R)”). This statement is a revision of
SFAS No. 123, and supersedes APB Opinion No. 25,
and its related implementation guidance. SFAS 123(R)
addresses all forms of share based payment (“SBP”)
awards including shares issued under employee stock purchase
plans, stock options, restricted stock and stock appreciation
rights. Under SFAS 123(R), SBP awards result in a cost that
will be measured at fair value on the awards’ grant date,
based on the estimated number of awards that are expected to
vest and will result in a charge to operations.
The Company had a choice of two attribution methods for
allocating compensation costs under SFAS 123(R): the
“straight-line method,” which allocates expense on a
straight-line basis over the requisite service period of the
last separately vesting portion of an award, or the “graded
vesting attribution method,” which allocates expense on a
straight-line basis over the requisite service period for each
separately vesting portion of the award as if the award was, in
substance, multiple awards. The Company chose the former method
and amortized the fair value of each option on a straight-line
basis over the requisite period of the last separately vesting
portion of each award.
73
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Under SFAS 123(R), nonpublic entities, including those that
become public entities after June 15, 2005, that used the
minimum value method of measuring equity share options and
similar instruments for either recognition or pro forma
disclosure purposes under SFAS No. 123 are required to
apply SFAS 123(R)prospectively to new awards and to awards
modified, repurchased, or cancelled after the date of adoption.
In addition, SFAS 123(R), requires such entities to
continue accounting for any portion of awards outstanding at the
date of initial application using the accounting principles
originally applied to those awards. Accordingly, stock-based
compensation expense relating to awards granted prior to
April 1, 2006 that are expected to vest in periods ending
after April 1, 2006 are being recorded in accordance with
the provisions of APB 25 and its related interpretive guidance.
The Company has adopted the prospective method with respect to
accounting for its transition to SFAS 123(R). Accordingly,
the Company recognized in salaries and related expense in the
accompanying consolidated statements of operations $158,000 of
stock-based compensation expense during the year ended
March 31, 2007, which represents the intrinsic value
amortization of options granted prior to April 1, 2006 that
the Company is continuing to account for using the recognition
and measurement principles prescribed under APB 25. The Company
also recognized in salaries and related expense in the
accompanying consolidated statements of operations $815,000 of
stock-based compensation expense during the year ended
March 31, 2007 which represents the amortization of the
fair value of options granted subsequent to adoption of
SFAS 123(R). During the year ended March 31, 2007, the
Company reclassified certain components of its
stockholders’ equity to reflect the elimination of deferred
compensation arising from unvested share-based compensation
pursuant to the requirements of Staff Accounting
Bulletin No. 107, regarding SFAS. 123(R). This
deferred compensation was previously recorded as an increase to
additional paid-in capital with a corresponding reduction to
stockholders’ equity for such deferred compensation. This
reclassification had no effect on net loss or total
stockholders’ equity (deficit) as previously reported. The
Company will record an increase to additional paid-in capital as
the share-based payments vest.
Non-Employee
Stock-Based Compensation
The Company accounts for equity instruments issued to
non-employees in accordance with the provisions of
SFAS No. 123(R) and EITF Issue
No. 96-18,
“Accounting for Equity Instruments That are Issued to Other
Than Employees for Acquiring, or in Conjunction with Selling,
Goods or Services,”
(“EITF 96-18”)
which requires that such equity instruments are recorded at
their fair value on the measurement date. The measurement of
stock-based compensation is subject to periodic adjustment as
the underlying equity instrument vests. Non-employee stock-based
compensation charges are being amortized over the vesting period.
Income
Taxes
The Company accounts for income taxes in accordance with
SFAS No. 109, Accounting for Income Taxes
(“SFAS No. 109”). Under
SFAS No. 109, deferred tax assets and liabilities are
determined based on the differences between the financial
reporting and tax bases of assets and liabilities and net
operating loss and credit carryforwards using enacted tax rates
in effect for the year in which the differences are expected to
impact taxable income. Valuation allowances are established when
necessary to reduce deferred tax assets to the amounts expected
to be realized.
Comprehensive
Loss
Other comprehensive loss includes all changes in
stockholders’ equity (deficit) during a period from
non-owner sources and is reported in the consolidated statement
of stockholders’ equity (deficit). To date, other
comprehensive loss consists of changes in accumulated foreign
currency translation adjustments during the years.
74
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Net
Loss Per Share
The Company computes net loss per share in accordance with
SFAS No. 128 “Earnings Per Share” and has
applied the guidance enumerated in Staff Accounting
Bulletin No. 98 (“SAB Topic 4D”) with
respect to evaluating its issuances of equity securities during
all periods presented.
Under SFAS No. 128, basic net loss per share is
computed by dividing net loss per share available to common
stockholders by the weighted average number of common shares
outstanding for the period and excludes the effects of any
potentially dilutive securities. Diluted earnings per share, if
presented, would include the dilution that would occur upon the
exercise or conversion of all potentially dilutive securities
into common stock using the “treasury stock”
and/or
“if converted” methods as applicable. The computation
of basic loss per share for the years ended March 31, 2007,
2006 and 2005, excludes potentially dilutive securities because
their inclusion would be anti-dilutive.
In addition to the above, the Securities and Exchange Commission
(“SEC”) (under SAB Topic 4D) requires new
registrants to retroactively include the dilutive effect of
common stock or potential common stock issued for nominal
consideration during all periods presented in its computation of
basic earnings (loss) per share and diluted earnings per share
as if they were, in substance, recapitalizations. The Company
evaluated all of its issuances of equity securities prior to the
completion of its IPO on January 30, 2007
(Note 13) and determined that it had no nominal
issuances of common stock or common stock equivalents to include
in its computation of loss per share for any of the years
presented.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
(In thousands)
|
|
|
|
|
Anti-dilutive securities
excluded from the computation of basic and diluted net loss per
share are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options to purchase common stock
|
|
|
2,020
|
|
|
|
1,969
|
|
|
|
1,340
|
|
|
Warrants to purchase common stock
|
|
|
1,369
|
|
|
|
858
|
|
|
|
464
|
|
|
Convertible preferred stock (as if
converted)
|
|
|
—
|
|
|
|
3,984
|
|
|
|
2,352
|
|
|
Warrants to purchase convertible
preferred stock (as if converted)
|
|
|
—
|
|
|
|
17
|
|
|
|
17
|
|
|
Convertible debt
|
|
|
—
|
|
|
|
—
|
|
|
|
10
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,389
|
|
|
|
6,828
|
|
|
|
4,183
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
During the year ended March 31, 2007, the Company issued
common stock in connection with the conversion of its
convertible preferred stock to common stock at the close of its
initial public offering, sold common stock in its initial public
offering, sold common stock in connection with its
underwriter’s partial exercise of their over- allotment
option and issued common stock in connection with the exercise
of warrants (Note 13). These transactions resulted in
significant additional dilution.
Fair
Value of Financial Instruments
The carrying amounts reported in the balance sheet for cash and
cash equivalents, accounts receivable, accounts payable and
accrued expenses approximate fair value based on the short-term
maturity of these instruments. The carrying amounts of the
Company’s line of credit obligation and other long term
obligations approximate fair value as such instruments feature
contractual interest rates that are consistent with current
market rates of interest or have effective yields that are
consistent with instruments of similar risk, when taken together
with equity instruments issued to the holder.
75
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Preferred
Stock
The Company applies the guidance enumerated in
SFAS No. 150 “Accounting for Certain Financial
Instruments with Characteristics of both Liabilities and
Equity” and EITF Topic D-98 “Classification and
Measurement of Redeemable Securities,” when determining the
classification and measurement of its convertible preferred
shares. Preferred shares subject to mandatory redemption (if
any) are classified as liability instruments and are measured at
fair value in accordance with SFAS 150. All other issuances
of preferred stock are subject to the classification and
measurement principles of EITF Topic D-98. Accordingly the
Company classifies conditionally redeemable preferred shares (if
any), which includes preferred shares that feature redemption
rights that are either within the control of the holder or
subject to redemption upon the occurrence of uncertain events
not solely within the Company’s control, as temporary
equity. At all other times, the Company classifies its preferred
shares in stockholders’ equity.
The Company’s convertible preferred shares did not feature
any redemption rights within the holder’s control or
conditional redemption features not within the Company’s
control as of March 31, 2006 and 2005. Accordingly all
issuances of convertible preferred stock are presented as a
component of stockholders equity (deficit). All shares of
convertible preferred stock automatically converted into to
common stock at the closing of the Company’s initial public
offering on January 30, 2007.
Convertible
Instruments
The Company evaluates and accounts for conversion options
embedded in its convertible instruments in accordance with
SFAS No. 133 “Accounting for Derivative
Instruments and Hedging Activities”
(“SFAS 133”) and
EITF 00-19
“Accounting for Derivative Financial Instruments Indexed
to, and Potentially Settled in, a Company’s Own Stock”
(“EITF 00-19”).
SFAS 133 generally provides three criteria that, if met,
require companies to bifurcate conversion options from their
host instruments and account for them as free standing
derivative financial instruments in accordance with
EITF 00-19.
These three criteria include circumstances in which (a) the
economic characteristics and risks of the embedded derivative
instrument are not clearly and closely related to the economic
characteristics and risks of the host contract, (b) the
hybrid instrument that embodies both the embedded derivative
instrument and the host contract is not remeasured at fair value
under otherwise applicable generally accepted accounting
principles with changes in fair value reported in earnings as
they occur and (c) a separate instrument with the same
terms as the embedded derivative instrument would be considered
a derivative instrument subject to the requirements of
SFAS 133. SFAS 133 and
EITF 00-19
also provide an exception to this rule when the host instrument
is deemed to be conventional (as that term is described in the
implementation guidance to SFAS 133 and further clarified
in
EITF 05-2
“The Meaning of “Conventional Convertible Debt
Instrument” in Issue
No. 00-19).
The Company accounts for convertible instruments (when it has
determined that the embedded conversion options should not be
bifurcated from their host instruments) in accordance with the
provisions of
EITF 98-5
“Accounting for Convertible Securities with Beneficial
Conversion Features,”
(“EITF 98-5”)
and
EITF 00-27
“Application of
EITF 98-5
to Certain Convertible Instruments.” Accordingly, the
Company records when necessary discounts to convertible notes
for the intrinsic value of conversion options embedded in debt
instruments based upon the differences between the fair value of
the underlying common stock at the commitment date of the note
transaction and the effective conversion price embedded in the
note. Debt discounts under these arrangements are amortized over
the term of the related debt to their earliest date of
redemption. The Company also records when necessary deemed
dividends for the intrinsic value of conversion options embedded
in preferred shares based upon the differences between the fair
value of the underlying common stock at the commitment date of
the note transaction and the effective conversion price embedded
in the note.
The Company evaluated the conversion option embedded in its
convertible instruments during each of the reporting periods
presented and has determined, in accordance with the provisions
of these statements, that it does
76
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
not meet the criteria requiring bifurcation of these
instruments. Additionally, the Company’s conversion
options, if free standing, would not be considered derivatives
subject to accounting guidelines prescribed under SFAS 133.
The characteristics of common stock that is issuable upon a
holder’s exercise of conversion options embedded in the
Company’s preferred shares are deemed to be clearly and
closely related to the characteristics of the preferred shares
(as that term is clarified in paragraph 61 l of the
implementation guidance included in Appendix A of
SFAS 133). The Company did not record deemed dividends
during any of the periods presented because the effective
conversion price of the convertible preferred shares exceeded
the fair value of the Company common stock at the respective
dates of issuance.
Common
Stock Purchase Warrants and Other Derivative Financial
Instruments
The Company accounts for the issuance of common stock purchase
warrants issued and other free standing derivative financial
instruments in accordance with the provisions of
EITF 00-19.
Based on the provisions of
EITF 00-19,
the Company classifies as equity any contracts that
(i) require physical settlement or net-share settlement or
(ii) gives the Company a choice of net-cash settlement or
settlement in its own shares (physical settlement or net-share
settlement). The Company classifies as assets or liabilities any
contracts that (i) require net-cash settlement (including a
requirement to net cash settle the contract if an event occurs
and if that event is outside the control of the Company) or
(ii) gives the counterparty a choice of net-cash settlement
or settlement in shares (physical settlement or net-share
settlement). The Company assesses classification of its
freestanding derivatives at each reporting date to determine
whether a change in classification between assets and
liabilities is required. The Company determined that its
freestanding derivatives, which principally consists of warrants
to purchase common stock satisfied the criteria for
classification as equity instruments at March 31, 2007 and
2006.
Recent
Accounting Pronouncements
In March 2006, the FASB issued SFAS No. 156
“Accounting for Servicing of Financial Assets —
an amendment of FASB Statement No. 140”
(“SFAS 156”). SFAS 156 is effective for the
first fiscal year beginning after September 15, 2006.
SFAS 156 changes the way entities account for servicing
assets and obligations associated with financial assets acquired
or disposed of. The Company has not yet completed its evaluation
of the impact of adopting SFAS 156 on its results of
consolidated operations or financial position, but does not
expect that the adoption of SFAS 156 will have a material
impact.
In June 2006, the Financial Accounting Standards Board issued
FASB Interpretation No. 48, Accounting for Uncertainty in
Income Taxes. This Interpretation prescribes a recognition
threshold and measurement attribute for the financial statement
recognition and measurement of a tax position taken or expected
to be taken in a tax return. This Interpretation also provides
guidance on derecognition, classification, interest and
penalties, accounting in interim periods, disclosure, and
transition. The Interpretation is effective for fiscal years
beginning after December 15, 2006. The Company has not yet
completed its analysis of the impact this Interpretation will
have on its financial condition, results of operations, cash
flows or disclosures.
In February 2007, the FASB issued SFAS 159, “The Fair
Value Option for Financial Assets and Financial
Liabilities” (“SFAS 159”). SFAS 159
provides companies with an option to report selected financial
assets and liabilities at fair value and establishes
presentation and disclosure requirements designed to facilitate
comparisons between companies that choose different measurement
attributes for similar types of assets and liabilities.
SFAS 159 is effective for fiscal years beginning after
November 15, 2007. The Company is in the process of
evaluating the impact of the adoption of this statement on the
Company’s results of operations and financial condition.
In September 2006, the FASB issued SFAS No. 157,
“Accounting for Fair Value Measurements”
(“SFAS 157”). SFAS 157 defines fair value,
and establishes a framework for measuring fair value in
generally accepted accounting principles and expands disclosure
about fair value measurements. SFAS 157 is effective for
77
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
financial statements issued subsequent to November 15,
2007. The Company does not expect that the adoption of this the
standard will have a material impact on its financial position,
results of operations or cash flows.
In September 2006, the SEC issued Staff Accounting
Bulletin 108, Considering the Effects on Prior Year
Misstatements when Quantifying Misstatements in Current Year
Financial Statements, (“SAB 108”). SAB 108
requires registrants to quantify errors using both the income
statement method (i.e. iron curtain method) and the rollover
method and requires adjustment if either method indicates a
material error. If a correction in the current year relating to
prior year errors is material to the current year, then the
prior year financial information needs to be corrected. A
correction to the prior year results that are not material to
those years, would not require a restatement process where prior
financials would be amended. SAB 108 is effective for
fiscal years ending after November 15, 2006. The
Company’s adoption of SAB 108 did not have a material
effect on its financial position, results of operations or cash
flows.
In December 2006, the FASB issued FSP
EITF 00-19-2
“Accounting for Registration Payment Arrangements”
(“FSP
EITF 00-19-2”).
FSP
EITF 00-19-2
addresses an issuer’s accounting for registration payment
arrangements. This pronouncement specifies that the contingent
obligation to make future payments or otherwise transfer
consideration under a registration payment arrangement, whether
issued as a separate agreement or included as a provision of a
financial instrument, should be separately recognized and
accounted for as a contingency in accordance with SFAS 5
“Accounting for Contingencies.” FSP
EITF 00-19-2
amended previous standards relating to rights agreements became
effective on December 21, 2006 with respect to arrangements
entered into or modified beginning on such date and for the
first fiscal year beginning after December 15, 2006 with
respect to those arrangements entered into prior to
December 21, 2006. The Company is in the process of
evaluating the impact of the adoption of this statement on the
Company’s results of operations and financial condition.
Other accounting standards that have been issued or proposed by
the FASB or other standards-setting bodies that do not require
adoption until a future date are not expected to have a material
impact on the consolidated financial statements upon adoption.
|
|
|
NOTE 4
|
—
Accounts Receivable
|
Accounts receivable consists of the following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Accounts receivable
|
|
$
|
1,571
|
|
|
$
|
1,166
|
|
|
Less: allowance for doubtful
accounts
|
|
|
(207
|
)
|
|
|
(90
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,364
|
|
|
$
|
1,076
|
|
|
|
|
|
|
|
|
|
|
|
Inventories consist of the following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Raw materials
|
|
$
|
311
|
|
|
$
|
267
|
|
|
Finished goods
|
|
|
65
|
|
|
|
1,046
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
376
|
|
|
|
1,313
|
|
|
Less: inventory allowances
|
|
|
(94
|
)
|
|
|
(996
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
282
|
|
|
$
|
317
|
|
|
|
|
|
|
|
|
|
|
|
78
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
NOTE 6 —
|
Prepaid
Expenses and Other Current Assets
|
Prepaid expenses and other current assets consist of the
following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Prepaid expenses
|
|
$
|
976
|
|
|
$
|
304
|
|
|
Value Added Tax receivable
|
|
|
125
|
|
|
|
722
|
|
|
Other current assets
|
|
|
71
|
|
|
|
360
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,172
|
|
|
$
|
1,386
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NOTE 7 —
|
Debt
Issue Costs
|
Debt issue costs consists of the following (in thousands):
| |
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
|
|
Fair value of common stock
purchase warrants issued to Western Technologies, Inc. in
connection with a Line of Credit (Note 10)
|
|
$
|
1,046
|
|
|
Fair value of common stock
purchase warrants issued to Brookstreet Securities Corporation
(“Brookstreet”) in connection with a Bridge Loan
(Note 10)
|
|
|
105
|
|
|
Cash paid for debt offering
expenses
|
|
|
77
|
|
|
|
|
|
|
|
|
|
|
|
1,228
|
|
|
Less: accumulated amortization
recorded as non-cash interest expense
|
|
|
(402
|
)
|
|
|
|
|
|
|
|
|
|
$
|
826
|
|
|
|
|
|
|
|
|
|
|
NOTE 8 —
|
Property
and Equipment
|
Property and equipment consists of the following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Manufacturing, lab, and other
equipment
|
|
$
|
2,489
|
|
|
$
|
1,866
|
|
|
Office equipment
|
|
|
716
|
|
|
|
653
|
|
|
Furniture and fixtures
|
|
|
219
|
|
|
|
209
|
|
|
Leasehold improvements
|
|
|
489
|
|
|
|
498
|
|
|
Capital projects in progress
|
|
|
249
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,162
|
|
|
|
3,226
|
|
|
Less: accumulated depreciation and
amortization
|
|
|
(1,955
|
)
|
|
|
(1,286
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,207
|
|
|
$
|
1,940
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment includes $186,000 of equipment purchases
that were financed under capital lease obligations as of
March 31, 2007 and 2006 (Note 11). The accumulated
amortization on these assets amounted to $146,000 and $108,000
as of March 31, 2007 and 2006, respectively.
Depreciation expense (including amortization of leased assets)
amounted to $672,000, $651,000 and $434,000 for the years ended
March 31, 2007, 2006 and 2005, respectively.
79
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
NOTE 9 —
|
Accrued
Expenses and Other Current Liabilities
|
Accrued expenses and other current liabilities consist of the
following (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Salaries and related costs
|
|
$
|
525
|
|
|
$
|
267
|
|
|
Professional fees
|
|
|
524
|
|
|
|
673
|
|
|
Estimated liability for pending
litigation (Note 12)
|
|
|
21
|
|
|
|
300
|
|
|
Value Added Tax payable
|
|
|
—
|
|
|
|
220
|
|
|
Deferred revenue
|
|
|
55
|
|
|
|
156
|
|
|
Other
|
|
|
296
|
|
|
|
70
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,421
|
|
|
$
|
1,686
|
|
|
|
|
|
|
|
|
|
|
|
On May 1, 1999, the Company issued a note payable in the
amount of $64,000 with interest at 8% per annum and a final
payment due on December 31, 2009. The remaining balance on
this obligation, which amounts to $23,000 including accrued
interest, is included in non-current portion of long-term debt
in the accompanying consolidated balance sheet at March 31,
2007. During the year ended March 31, 2007, the Company
made principal payments on this note in the amount of $45,000.
During March 2004, the Company entered into an equipment
financing facility providing it with up to $1,000,000 of
available credit to finance equipment purchases through
March 31, 2005. During the year ended March 31, 2005,
the Company drew an aggregate of $994,000 of advances under this
facility, which are payable in 33 monthly installments with
interest at the rate of 13.5% per annum and mature at various
times through May 1, 2007. The Company also paid
approximately $82,000 of fees to the lender under this
arrangement including $5,000 in cash and $77,000 representing
the fair value of warrants to purchase up to 16,666 shares
of the Company’s Series A convertible preferred stock
which automatically converted to warrants to purchase common
stock at the closing of the Company’s initial public
offering on January 30, 2007 (Note 13). The Company
recorded the fair value of warrants and other fees as interest
expense during the year ended March 31, 2005, the period in
which the Company was permitted to draw advances under this
facility. All borrowings under this arrangement are
collateralized by the equipment financed under this facility.
The Company made principal payments on these notes which
amounted to $332,000, $337,000, and $305,000 during the years
ending March 31, 2007 and 2006 respectively. Interest
expense under these obligations amounted to $25,000, $73,000 and
$83,000 for the years ended March 31, 2007, 2006, and 2005,
respectively. The remaining principal balance amounted to
$19,000 at March 31, 2007, and is included in the current
portion of notes payable obligations in the accompanying
consolidated balance sheet.
From February 2005 to March 2006, the Company issued various
notes for aggregate principal amounting to $182,000 with
interest rates ranging from 6.25% to 14.44% per annum. The
proceeds of these notes were used to purchase automobiles and
software. The Company made principal payments on these notes of
$33,600 and $24,000, during the years ended March 31, 2007
and 2006, respectively. Aggregate interest expense under these
obligations amounted to $11,000, $8,900 and $1,000 for the years
ended March 31, 2007, 2006 and 2005, respectively. These
notes are payable in aggregate monthly installments of $3,700
including interest through March 14, 2011. The remaining
balance of these notes amounted to $123,000 at March 31,
2007, including $36,000 in the current portion of long-term debt
in the accompanying consolidated balance sheet.
On June 14, 2006, the Company entered into a credit
facility providing it with up to $5,000,000 of available credit.
The facility permitted the Company to borrow up to a maximum of
$2,750,000 for growth capital, $1,250,000 for working capital
based on eligible accounts receivable and $1,000,000 in
equipment financing. In
80
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
June 2006, the Company drew an aggregate of $4,182,000 of
borrowings under this facility. These borrowings are payable in
30 to 33 fixed monthly installments with interest at rates
ranging from 12.4% to 12.7% per annum, maturing at various times
through April 9, 2009. The Company has no unused
availability under this credit facility since amounts drawn
under the working capital facility were based upon an initial
measurement of eligible accounts receivable.
The Company also issued to the lender warrants to purchase up to
71,521 shares of its Series B convertible preferred
stock upon originating the loan which automatically converted
into warrants to purchase 71,534 shares of the
Company’s common stock at the closing of the Company’s
initial public offering on January 30, 2007. The aggregate
fair value of all warrants issued to the lender under this
arrangement amounts to $1,046,000 (Note 13). This amount
was recorded as debt issue costs in the March 31, 2007
consolidated balance sheet and is being amortized as interest
expense over the term of the credit facility or 30 to
33 months.
Borrowings under the growth capital line are collateralized by
the total assets of the Company. Borrowings under the equipment
line are collateralized by the underlying assets funded, and
borrowings under the working capital line are collateralized by
eligible accounts receivable. On a monthly basis, the Company
must maintain a 1:1 ratio of borrowing under the working
capital line to eligible accounts receivable. The Company has
30 days from each measurement date to either increase
eligible accounts receivable or pay the excess principal in the
event that the ratio is less than 1:1. No restrictive covenants
exist for either the equipment line or the growth capital line.
The Company is not required to direct customer remittances to a
lock box, nor does the credit agreement provide for subjective
acceleration of the loans. In connection with these notes, for
the year ended March 31, 2007, the Company made $852,000 of
principal payments, $333,000 of interest payments and recorded
$340,000 of non-cash interest expense related to the
amortization of debt issue costs. The aggregate remaining
principal balance under this facility amounted to $3,329,000,
including $1,500,000 in the current portion of long term debt in
the accompanying consolidated balance sheet at March 31,
2007. As of March 31, 2007, the Company no longer had the
ability to draw additional funds on the various lines.
On March 29, 2007, the Company entered into Amendment
No. 1 to the loan agreement described above. Pursuant to
the amendment the lender and the Company agreed that the
security interest in the Company’s intellectual property
would be removed and the lender’s security interest in the
Company’s assets would not include the Company’s
intellectual property unless and until the Company’s cash
and cash equivalents fall below 600% of the Company’s
average monthly expenses less non-cash charges. At
March 31, 2007, the Company’s cash and cash
equivalents position was in excess of 600% of its average
monthly expenses and therefore no lien against its intellectual
property was in place.
On May 5, 2006, the Company entered into a note agreement
for $69,000 with interest at the rate of 7.94% percent per
annum. The proceeds of this note were used to purchase an
automobile. This note is payable in monthly installments of
$1,200 through May 2012. The Company made principal payments of
$7,400 and interest payments of $5,000 during the year ended
March 31, 2007. The remaining balance of this note amounted
to $61,000 at March 31, 2007, including $9,800 in the
current portion of long-term debt in the accompanying
consolidated balance sheet.
From July 1, 2006 to March 25, 2007, the Company
entered into note agreements for $805,000 with interest rates
ranging from 7.0% to 9.7% per annum. The proceeds of these notes
were used to finance insurance premiums. The remaining balance
of these notes are payable in aggregate monthly installments of
$66,000 through November 25, 2007. The Company made
principal payments of $464,000 and interest payments of $10,500
during the year ended March 31, 2007. The remaining balance
of these notes amounted to $480,000 at March 31, 2007, and
is included in the current portion of long-term debt in the
accompanying consolidated balance sheet.
On November 7, 2006, the Company signed a loan agreement
with Robert Burlingame, one of the Company’s directors, in
the amount of $4,000,000, which was funded on November 10,
2006 and which accrues interest at an annual rate of 7%.
Concurrently, Mr. Burlingame became a consultant to the
Company under a two-year consulting
81
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
agreement, and was appointed to fill a vacancy on the
Company’s Board of Directors. The principal and all accrued
interest under the loan agreement will become due and payable in
full with interest on November 10, 2007. The loan is
secured by all assets of the Company, other than intellectual
property, and is subordinate to the security interest held by
the Company’s secured lender. At the time the principal was
advanced to the Company, Brookstreet, who acted as the agent in
this transaction, was paid a fee of $50,000 and was granted a
warrant to purchase 25,000 shares of the Company’s
common stock at an exercise price of $18.00 per share. The
aggregate fair value of all warrants issued to the agent under
this arrangement amounts to $105,000 (Note 13). This amount
in addition to the $50,000 cash payment was recorded as debt
issue costs in the March 31, 2007 consolidated balance
sheet and is being amortized as interest expense over the term
of the credit facility. During the year ended March 31,
2007, the Company recorded $62,000 of non-cash interest expense
related to the amortization of the debt issue costs and recorded
$109,000 of accrued interest expense related to this note. The
$4,000,000 loan is included in the current portion of long-term
debt in the accompanying consolidated balance sheet at
March 31, 2007.
On March 29, 2007, the Company entered into Amendment
No. 1 to the loan agreement described above. Pursuant to
the Amendment, the Company will make monthly interest payments
on the $4,000,000 principal of the original promissory note and
deposited $2,000,000 into a segregated interest-bearing account.
Under a second Amendment No. 2 to the loan agreement, Mr.
Burlingame has been granted sole signatory rights on this
account but may not make any draws on the account until the loan
obligation matures. The Company also agreed to deposit an
additional $2,000,000 into this account if its cash and cash
equivalents drop below $10,000,000 (including the amounts in
this account). The Company may withdraw accrued interest from
this account at any time, but has agreed not to withdraw
principal amounts from this account without the prior consent of
Mr. Burlingame. The Company has agreed that if it receives
unrestricted funds from the issuance of debt, or equity funding,
in excess of $500,000 prior to November 7, 2007, it will
pay such amounts to Mr. Burlingame to reduce the amounts
owing under the note.
A summary of principal payments due in years subsequent to
March 31, 2007 is as follows (in thousands):
| |
|
|
|
|
|
For Years Ending March 31,
|
|
|
|
|
|
|
2008
|
|
$
|
6,045
|
|
|
2009
|
|
|
1,836
|
|
|
2010
|
|
|
116
|
|
|
2011
|
|
|
23
|
|
|
2012
|
|
|
15
|
|
|
|
|
|
|
|
|
Total principal payments
|
|
|
8,035
|
|
|
Less: current portion
|
|
|
6,045
|
|
|
|
|
|
|
|
|
Long-term portion
|
|
$
|
1,990
|
|
|
|
|
|
|
|
|
|
|
NOTE 11 —
|
Capital
Lease Obligations
|
During the period of September 1, 2003 through
October 1, 2003, the Company entered into various capital
leases under which the aggregate present value of the minimum
lease payments amounted to $40,000. The present value of the
minimum lease payments was calculated using discount rates of
ranging from 13% to 18%. Lease payments, including amounts
representing interest, amounted to $12,000, $11,000 and $11,000
for the years ended March 31, 2007, 2006 and 2005,
respectively. The remaining principal balance on these
obligations amounted to $19,700 at March 31, 2007,
including $9,100 included in the current portion of capital
lease obligations in the accompanying consolidated balance sheet.
On November 10, 2004, the Company entered into a capital
lease under which the present value of the minimum lease
payments amounted to $37,000. The present value of the minimum
lease payments was calculated using a discount rate of 10%.
Lease payments, including amounts representing interest,
amounted to $9,300, $8,500
82
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
and $3,900 for the years ended March 31, 2007, 2006 and
2005, respectively. The remaining principal balance on these
obligations amounted to $21,900 at March 31, 2007,
including $7,500 included in the current portion of capital
lease obligations in the accompanying consolidated balance sheet.
The Company recorded interest expense in connection with these
lease agreements in the amounts of $6,700, $8,900 and $11,000
for the years ended March 31, 2007, 2006 and 2005,
respectively.
Minimum lease payments due in years subsequent to March 31,
2007 are as follows (in thousands):
| |
|
|
|
|
|
For Years Ending March 31,
|
|
|
|
|
|
|
2008
|
|
$
|
21
|
|
|
2009
|
|
|
21
|
|
|
2010
|
|
|
6
|
|
|
Total minimum lease payments
|
|
|
48
|
|
|
Less: amounts representing interest
|
|
|
6
|
|
|
|
|
|
|
|
|
Present value of minimum lease
payments
|
|
|
42
|
|
|
Less: current portion
|
|
|
17
|
|
|
|
|
|
|
|
|
Long-term portion
|
|
$
|
25
|
|
|
|
|
|
|
|
|
|
|
NOTE 12 —
|
Commitments
and Contingencies
|
Lease
Commitments
The Company has entered into various non-cancelable operating
leases, primarily for office facility space, that expire at
various times through April 2012. Minimum lease payments for
non-cancelable operating leases are as follows (in thousands):
| |
|
|
|
|
|
For Years Ending March 31,
|
|
|
|
|
|
|
2008
|
|
$
|
296
|
|
|
2009
|
|
|
170
|
|
|
2010
|
|
|
91
|
|
|
2011
|
|
|
95
|
|
|
2012
|
|
|
8
|
|
|
|
|
|
|
|
|
Total minimum lease payments
|
|
$
|
660
|
|
|
|
|
|
|
|
Rent expense amounted to $590,000, $535,000 and $510,000 for the
years ended March 31, 2007, 2006, and 2005, respectively.
Employment
Agreements
The Company entered into employment agreements with five of its
key executives. The agreements provide, among other things, for
the payment of aggregate annual salaries of approximately
$915,000 and twelve to twenty four months of severance
compensation for terminations under certain circumstances.
Aggregate potential severance compensation amounted to
$1,275,000 at March 31, 2007.
On October 1, 2005, the Board authorized the grant to one
of the Company’s officers at the closing of its initial
public offering of an option under the 2004 Stock Option Plan to
purchase 60,000 shares of the Company’s common stock
at an exercise price of $3.00 per share. Due to the Board’s
subsequent decision not to grant additional options under the
2004 Stock Option Plan, the adoption by the Board and approval
by the stockholders of the Company’s 2006 Stock Incentive
Plan, and certain regulatory developments, on April 26,
2007, in lieu of the award of an
83
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
option, the Compensation Committee authorized the award to the
officer of 60,000 stock units (Note 20). Following the
measurement and expense recognition provisions enumerated in
SFAS 123(R), the Company recorded $351,000 of stock-based
compensation related to this award which is included in the
accompanying consolidated statement of operations for the year
ended March 31, 2007.
Legal
Matters
In April 2005, the Company was named as a defendant in an
employment related matter under a complaint filed by one of its
former employees in the Superior Court of the State of
California, in the County of Sonoma. The Company entered into a
settlement agreement with the plaintiff in November 2006, which
provided for the payment of $250,000 and the issuance of a
warrant to purchase 50,000 shares of the Company’s
common stock exercisable at $3.00 per share. The warrants, which
were non-forfeitable at the date of issuance, were recorded at
fair value which resulted in a $365,000 charge which was
recorded as a selling, general and administrative expense. A
$300,000 reserve was established and recorded in accrued
expenses in the accompanying balance sheet at March 31,
2006 which at the time was the Company’s best estimate of
the potential loss. The $250,000 cash payment was made in
February 2007. The issuance of the warrants was subject to the
Company obtaining appropriate waivers from the Company’s
convertible preferred stockholders which was obtained in
December 2006. Under the terms of the agreement, the plaintiff
has agreed to dismiss his claim and has waived any other
previous claims against the Company.
In November 2005, the Company identified a possible criminal
misappropriation of its technology in Mexico, and it notified
the Mexican Attorney General’s office. The Company believes
the Mexican Attorney General is currently conducting an
investigation.
On March 14, 2006, the Company filed suit in the
U.S. District Court for the Northern District of California
against Nofil Corporation and Naoshi Kono, Chief Executive
Officer of Nofil, for breach of contract, misappropriation of
trade secrets and trademark infringement. The Company believes
that Nofil Corporation violated key terms of both an exclusive
purchase agreement and non-disclosure agreement by contacting
and working with a potential competitor in Mexico. In the
complaint, the Company seeks damages of $3,500,000 and immediate
injunctive relief. On February 13, 2007, the Company
received the defendant’s answer and cross-complaint. The
cross-complaint, which alleges fraudulent inducement to enter
contracts, breach of non-disclosure contract, trade secret
misappropriation, conversion and violation under civil RICO
statutes by the Company, seeks damages in excess of $4,500,000.
The Company believes that the cross-complaint, and allegations
therein, are without merit. No trial date has been set. The
Company cannot predict the outcome of this matter nor can it
estimate a range of possible loss. While the Company does not
anticipate that the outcome of this matter is likely to result
in a material loss, there can be no assurance that such outcome
will not have a material adverse effect on the Company’s
financial condition or results of operations.
The Company is currently in discussions regarding two trademark
matters asserting confusion in trademarks with respect to the
Company’s use of the name Microcyn60 in Mexico. Although
the Company believes that the nature and intended use of its
products are different from those with the similar names, it has
agreed with one of the parties to stop using the name Microcyn60
by September 2007. Although such plaintiff referred the matter
to the Mexico Trademark Office, the Company is not aware of a
claim for monetary damages. The Company is in discussions with
the other party and believes that the name change will satisfy
an assertion of confusion; however, Company management believes
that the Company could incur a possible loss of approximately
$100,000 for the use of the name Microcyn60 during the twelve
month period following the date of settlement.
In June 2006, the Company received a written communication from
the grantor of a license to an earlier version of its technology
indicating that such license was terminated due to an alleged
breach of the license agreement by the Company. The license
agreement extends to the Company’s use of the technology in
Japan only. While the Company does not believe that the
grantor’s revocation is valid under the terms of the
license agreement
84
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
and no legal claim has been threatened to date, the Company
cannot provide any assurance that the grantor will not take
legal action to restrict the Company’s use of the
technology in the licensed territory.
While Company management does not anticipate that the outcome of
this matter is likely to result in a material loss, there can be
no assurance that if the grantor pursues legal action, such
legal action would not have a material adverse effect on the
Company’s financial position or results of operations.
In August 2006, the Company received a “show cause”
letter from the U.S. Environmental Protection Agency
(“EPA”), which stated that, in tests conducted by the
EPA, Cidalcyn was found to be ineffective in killing certain
specified pathogens when used according to label directions.
Based on its results, the EPA strongly recommended that the
Company immediately recalled all Cidalcyn distributed on and
after September 28, 2005. Accordingly, the Company
commenced a voluntary recall of Cidalcyn. Although the Company
has not marketed Cidalcyn on a large commercial scale, it has
provided it in small quantities to numerous hospitals solely for
use in product evaluation exercises. In a second letter, the EPA
stated it intended to file a civil administrative complaint
against the Company for violation of the Federal Insecticide,
Fungicide, and Rodenticide Act (“FIFRA”). Under FIFRA,
the EPA could assess civil penalties related to the sale and
distribution of a pesticide product not meeting the label’s
claims as a broad-spectrum hospital disinfectant. In April 2007,
the Company paid a settlement amounting to $20,800 to the EPA in
connection with this matter.
In September 2006, a consulting firm in Mexico City contacted
the Company threatening legal action in Mexico, alleging breach
of contract and claiming damages of $225,000. A formal compliant
was not served. In December 2006, the Company entered into a
settlement agreement with the consulting firm where the Company
paid $115,000 for the dismissal of their claim and waiver of any
previous claims against the Company.
In February 2007, the Company’s Mexico subsidiary served
Quimica Pasteur (“QP”), a former distributor of the
Company’s products in Mexico, with a claim alleging breach
of contract under a note made by QP. A trial date has not yet
been set.
The Company, from time to time, is involved in legal matters
arising in the ordinary course of its business. While management
believes that such matters are currently insignificant, there
can be no assurance that matters arising in the ordinary course
of business for which the Company is or could become involved in
litigation, will not have a material adverse effect on its
business, financial condition or results of operations.
Consulting
Agreements
On October 1, 2005, the Company entered into a consulting
agreement with White Moon Medical. Akihisa Akao, a member of the
Board of Directors, is the sole stockholder of White Moon
Medical. Under the terms of the agreement, the individual will
be compensated for services provided outside his normal Board
duties. The Company paid and recorded expense related to this
agreement in the amount of $146,000 and $73,000 which is
included in selling, general and administrative expense in the
consolidated statements of operations for the years ended
March 31, 2007 and 2006, respectively. During the year
ended March 31, 2007, the Company extended the agreement
for an additional one-year term and continued to make the
monthly payments.
On November 7, 2006, the Company entered into a consulting
agreement with Mr. Robert Burlingame, one of the
Company’s directors who also provided the Company with a
$4,000,000 Bridge Loan (Note 10). The director received
warrants in connection with this agreement. During the year
ended March 31, 2007, the amortized fair value of the
warrants amounted to $70,000 and was recorded as selling,
general and administrative expense in the accompanying
consolidated statements of operations (Note 13).
85
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
NOTE 13 —
|
Stockholders’
Equity
|
Authorized
Capital
The Company is authorized to issue up to 100,000,000 shares
of common stock and 30,000,000 shares of convertible
preferred stock of which 1,375,000 shares have been
designated as Series A convertible preferred stock,
2,805,555 shares have been designated as Series B
convertible preferred stock and 875,000 shares have been
designated Series C convertible preferred stock. As
described in Note 1, the Company reincorporated in Delaware
on December 15, 2006 and now has common stock, with a par
value of $0.0001 per share.
Description
of Common Stock
Each share of common stock has the right to one
vote. The holders of common stock are entitled to
dividends when funds are legally available and when declared by
the Board of Directors.
Convertible
Preferred Stock
At the close of the Company’s initial public offering on
January 30, 2007, all 4,176,498 outstanding shares of
Series A, Series B, and Series C convertible
preferred stock automatically converted into an equal number of
shares of common stock.
The Company issued in a private placement transaction an
aggregate of 2,635,744 shares of its Series B
convertible preferred stock for net proceeds of $43,722,000
(gross proceeds of $47,446,000 less offering costs of
$3,724,000) including 1,621,651 shares issued during the
year ended March 31, 2006 for net proceeds of $27,026,000
and 1,014,093 shares issued during the year ended
March 31, 2005 for net proceeds of $16,696,000.
The Company issued in a private placement transaction an
aggregate of 193,045 shares of its Series C
convertible preferred stock for net proceeds of $2,903,000
(gross proceeds of $3,474,000 less offering costs of $571,000)
during the year ended March 31, 2007.
The Company had reserved 5,055,555 shares of its common
stock for issuance upon the conversion of its convertible
preferred stock.
Prior to conversion, the Series A preferred shares were
convertible into common stock at any time, at the option of the
holder at a fixed conversion price of $6.00 per share. The
Series B and Series C convertible preferred shares
were convertible into common stock at any time, at the option of
the holder, at a fixed conversion price of $18.00 per share. The
conversion prices of the Series A, Series B and
Series C convertible preferred shares were subject to
adjustment for stock splits, stock dividends, recapitalizations,
dilutive issuances and other anti-dilution provisions, including
circumstances in which the Company, at its discretion, issued
equity securities or convertible instruments that featured
prices lower than the conversion prices specified in the
Series A, B and C convertible preferred shares. The
Series A, Series B and Series C convertible
preferred shares were also automatically convertible into shares
of the Company’s common stock, at the then applicable
conversion price, (i) in the event that the holders of
two-thirds of the outstanding shares of Series A,
Series B and Series C convertible preferred shares
consented to such conversion; or (ii) upon the closing of a
firm commitment underwritten public offering of shares of common
stock of the Company yielding aggregate proceeds of not less
than $20 million (before deduction of underwriters
commissions and expenses); or (iii) Company’s going
public by means of a merger or acquisition which had a resultant
market capitalization of greater than $75 million.
Each share of Series A, Series B and Series C
convertible preferred shares had voting rights equal to an
equivalent number of common shares into which it was convertible
and voted together as one class with common stock. The holders
of the Series A were entitled to receive cumulative
dividends in preference to any dividend on the common stock at
the rate of 6% per annum on the initial investment amount
commencing January 1, 2006 (discussion below). The Company
had the option of paying the dividend in either common stock or
cash. The holders of Series B were entitled to receive
non-cumulative dividends when and if declared by the Board. The
86
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
holders of Series C were entitled to non-cumulative
dividends when and if declared by the Board and only after the
Series A had been paid all accrued dividends and any
dividends declared by the Board and payable to the Series B
had been paid. The holders of Series A, Series B and
Series C were also entitled to participate pro rata in any
dividends paid on the common stock, if declared by the Board of
Directors on an as converted basis.
In the event of any liquidation or winding up of the Company,
the holders of the Series A would have been entitled to
participate in the ratable distribution of the assets of the
Company until the holders of the Series A had received a
per share amount equal to $12.00 plus any declared but unpaid
dividends. The holders of Series B were entitled to
participate in the ratable distribution of the assets of the
Company after the holders of Series A had received a per
share amount equal to $12.00 and holders of Series B had
received a per share amount equal to $22.50, plus any declared
but unpaid dividends. The holders of Series C were entitled
to participate in the ratable distribution of the assets of the
Company after the holders of Series A had received a per
share amount equal to $12.00, Series B had received a per
share amount equal to $22.50 and Series C had received a
per share amount equal to $22.50, plus any declared but unpaid
dividends. Thereafter, any remaining assets would have needed to
be distributed ratably to the holders of common stock until the
common stockholders had received a per share amount equal to
$12.00. Any remaining assets of the Company thereafter would
have been distributed ratably to the Series A convertible
preferred stockholders, Series B convertible preferred
stockholders, Series C convertible preferred stockholders
and to the common stockholders, on an as converted basis.
Liquidation events included (i) a final dissolution or
winding up of the Company’s affairs requiring a liquidation
of all classes of stock, (ii) a merger, consolidation or
similar event resulting in a more than 50% change in control,
(iii) the sale of all or substantially all of the
Company’s assets and (iv) the effectuation (at the
Company’s election) of any transaction or series of
transactions resulting in a more than 50% change in control.
Under the terms of Series A, Series B and
Series C investors rights agreements between the Company
and its convertible preferred stockholders, any time after
January 30, 2007 following the Company’s completion of
its IPO, the Series A, Series B and Series C
investors entitled to registration rights could request that the
Company file a registration statement covering the public sale
of the underlying common stock under the Securities Act of 1933,
as amended (the “1933 Act”) with limited rights
to delay by the Company. The investors having registration
rights are also entitled to unlimited piggyback registration
rights on all 1933 Act registrations of the Company (except
for registrations relating to employee benefit plans on
Form S-8
and corporate reorganizations on
Form S-4).
The foregoing demand and piggyback registration rights terminate
on the earlier of (i) one year after the Company’s
IPO, (ii) such time as Rule 144 or another similar
exemption under the 1933 Act is available for sale of all
of an investor’s shares during a three-month period without
registration or (iii) with respect to each investor, where
the investor no longer holds 1% the outstanding shares of the
Company. The investors rights agreement also placed certain
restrictions on the convertible preferred stockholders from
selling their shares and provided them with certain rights of
first refusal, co-sale and drag along and tag along rights for
sales effectuated under certain circumstances. These rights
expired upon the conversion of the preferred shares into common
stock.
As described in Note 3, the Company applied the
classification and measurement principles enumerated in EITF
Topic D-98 with respect to accounting for its issuances of the
Series A, Series B and Series C convertible
preferred stock. The Company was required, under California law,
to obtain the approval of its Board of Directors in order to
effectuate a merger, consolidation or similar event resulting in
a more than 50% change in control or a sale of all or
substantially all of its assets. The Board of Directors would
then be required to submit proposals to enter into these types
of transactions to its stockholders for their approval by
majority vote. At any reporting date, the Company’s
convertible preferred stockholders did not (i) have control
of the Company’s Board of Directors and (ii) did not
have sufficient voting rights to control a redemption of these
shares by either of these events. In addition the effectuation
of any transaction or series of transactions resulting in a more
than 50% change in control of the Company could be made only by
the Company at its own election. Based on these provisions, the
Company classified its Series A, Series B and
Series C convertible preferred shares in stockholders’
equity in the
87
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
accompanying March 31, 2007 consolidated balance sheet
because the liquidation events were deemed to be within the
Company’s control in accordance with the provisions of EITF
Topic D-98.
Also as described in Note 3, the Company evaluated the
conversion options embedded in the Series A, Series B
and Series C preferred shares to determine (in accordance
with SFAS 133 and
EITF 00-19)
whether they should have bifurcated them from their host
instruments and accounted for them as separate derivative
financial instruments. The Company determined, in accordance
with SFAS 133, that the risks and rewards of the common
shares underlying the conversion feature were clearly and
closely related to those of the host instrument. Accordingly the
conversion features, which were not deemed to be beneficial at
the commitment dates of these financing transactions, were
accounted for as embedded conversion options in accordance with
EITF 98-5
and
EITF 00-27.
The Company evaluated the Series A, Series B and
Series C preferred shares at each reporting date for
appropriate balance sheet classification. The Company concluded
the appropriate balance sheet classification for the convertible
preferred stock at each reporting date was in permanent equity
in the accompanying consolidated balance sheets at
March 31, 2007 and 2006.
Initial
Public Offering
The Company’s Registration Statement on
Form S-1,
Amendment No. 7, (File
No. 333-135584)
related to its IPO was declared effective by the SEC on
January 24, 2007. A total of 3,025,000 shares of the
Company’s common stock were registered with the SEC. All of
these shares were registered on the Company’s behalf. The
offering commenced on January 25, 2007 and
3,025,000 shares of common stock offered were sold on
January 30, 2007 for an aggregate offering price of
$24,200,000 through the managing underwriters: Roth Capital
Partners, Maxim LLC and Brookstreet Securities Corporation.
On February 16, 2007 the underwriters of the Company’s
initial public offering exercised a portion of their
over-allotment option and purchased 328,550 shares of the
Company’s common stock in accordance with the terms of the
underwriting agreement for an aggregate offering price of
$2,628,000 through the managing underwriters: Roth Capital
Partners, Maxim LLC and Brookstreet Securities Corporation.
The Company paid to the underwriters underwriting discounts,
commissions and non-accountable expenses totaling $2,146,000 in
connection with the initial public offering and the
underwriters’ exercise of the over-allotment shares. In
addition, the Company incurred additional expenses of
approximately $2,746,000 in connection with the initial public
offering, which when added to the underwriting discounts,
commissions and non-accountable expenses paid by the Company
amounts to total expenses of $4,892,000. Thus the net offering
proceeds to the Company (after deducting underwriting discounts
and commissions and offering expenses) were approximately
$21,936,000. No offering expenses were paid directly or
indirectly to any of the Company’s directors or officers
(or their associates), persons owning ten percent (10%) or more
of any class of the Company’s equity securities or to any
other affiliates.
Dividend
Payment in Common Stock
On February 15, 2007 the Board of Directors authorized
payment of dividends to the persons who were holders of the
Series A convertible preferred stock immediately prior to
the close of the IPO. The Company issued 87,494 shares of
common stock in payment of the dividend on March 21, 2007.
In connection with the accrued dividend, the Company’s net
loss available to common stockholders increased $404,000 and
$121,000 for the years ended March 31, 2007, and 2006,
respectively.
Stock
Purchase Warrants Issued in Financing Transactions
On October 27, 2005, the Company issued a warrant to
purchase 329,483 shares of common stock at an exercise
price of $18.00 per share to the placement agent that managed
the Series B stock offering. The warrants
88
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
were fully exercisable at the date of issuance with no future
performance obligations by the placement agent and expire the
second year following an IPO by the Company.
On June 14, 2006, the Company issued warrants to purchase
71,521 shares of Series B convertible preferred stock
at an exercise price of $18.00 per share in connection with the
new financing facility described in Note 10. These warrants
were automatically converted to warrants to purchase
71,521 shares of common stock at the closing of the
Company’s IPO on January 30, 2007. The warrants were
valued using the Black-Scholes pricing model. Assumptions used
were as follows: Fair value of the underlying stock $18.00;
risk-free interest rate 5.15% percent; contractual life of
11 years; dividend yield of 0%; and volatility of 70%. The
fair value of the warrants, which amounted to $1,046,000, was
recorded as deferred debt issue costs and is being amortized as
interest expense over the term of the credit facility.
Amortization of these costs amounted to $332,000 and is included
as a component of interest expense in the accompanying
consolidated statement of operations for the year ended
March 31, 2007.
On September 20, 2006 and October 14, 2006, the
Company issued a warrant to purchase 10,567 and
13,560 shares of common stock, respectively, at an exercise
price of $18.00 per share to the placement agent of the
Series C stock offering. The warrants were fully
exercisable at the date of issuance with no future performance
obligations by the placement agent and expire five years from
the date of issuance.
On September 20, 2006 and October 14, 2006 the Company
issued warrants to purchase 16,907 and 21,696 shares of
common stock, respectively, at an exercise price of $18.00 per
share to investors in conjunction with the purchase of
Series C stock units. The warrants require settlement in
shares of the Company’s common stock. The Company accounts
for the issuance of common stock purchase warrants issued in
connection with sales of its Units in accordance with the
provisions of
EITF 00-19
“Accounting for Derivative Financial Instruments Indexed
to, and Potentially Settled in, a Company’s Own
Stock”. Based on the provisions of
EITF 00-19,
the Company classified the warrants as equity. In addition, the
Company determined the convertible preferred stock was issued
with no effective beneficial conversion feature and therefore it
was not necessary to record a deemed dividend.
On November 10, 2006, Brookstreet Securities Corporation
was granted a warrant to purchase 25,000 shares of the
Company’s common stock at an exercise price of $18.00 per
share in connection with a finder’s fee for the Robert
Burlingame Bridge Loan, which funded on November 10, 2006
(Note 10). The warrants were valued using the Black-Scholes
pricing model. The fair value of the warrants, which amounted to
$105,000 and was recorded as debt issue costs in the
accompanying consolidated balance sheet as of March 31,
2007 and will be amortized as interest expense over the term of
the loan. The Company amortized $42,000 of interest expense
related to the warrants during the year ended March 31,
2007.
On January 30, 2007, under the terms of the Underwriting
Agreement and in connection with the closing of the
Company’s IPO, the Company issued to the underwriter’s
warrants to purchase an aggregate of 211,750 shares of
common stock at an exercise price of $13.20. On
February 16, 2007, under the terms of the Underwriting
Agreement and in connection with the closing of the partial
exercise of the underwriters’ over-allotment option, the
Company issued to the underwriters warrants to purchase an
aggregate of 22,998 shares of the common stock of the
Company at an exercise price of $13.20. The warrants were fully
exercisable at the date of issuance with no future performance
obligations by the underwriters and expire on January 29,
2012.
Common
Stock and Common Stock Purchase Warrants Issued to
Non-Employees
At various dates during the year ended March 31, 2006, the
Company issued warrants to purchase 73,843 shares of common
stock to various consultants at an exercise price of $18.00 per
share. Fair value of the underlying stock at the date of grant
was $10.16 per share. The warrants become exercisable at various
dates through November 11, 2009 and expire at various dates
through August 31, 2015. The fair value of the warrants
amounted to $158,000 and $153,000 and was recorded as a selling,
general and administrative expense in the accompanying
consolidated statement of operations for the year ended
March 31, 2007 and 2006, respectively.
89
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The non-vested warrants were adjusted to fair value at each
reporting date using the following weighted average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Fair market value of common stock
at reporting date
|
|
$
|
5.95
|
|
|
$
|
12.00
|
|
|
Estimated life
|
|
|
5.33 years
|
|
|
|
6.24 years
|
|
|
Risk-free interest rate
|
|
|
4.71
|
%
|
|
|
4.85
|
%
|
|
Dividend yield
|
|
|
0.00
|
%
|
|
|
0.00
|
%
|
|
Volatility
|
|
|
70
|
%
|
|
|
70
|
%
|
On November 10, 2006, the Company entered into a
2 year consulting agreement with its new director, Robert
Burlingame. Under the terms of the agreement, the Company issued
the director a warrant to purchase 75,000 shares of the
Company’s common stock, exercisable at a price equal to the
Company’s common stock in its initial public offering in
consideration of corporate advisory services. The warrants were
fully exercisable and non-forfeitable at date of issuance. The
warrants were valued using the Black-Scholes option pricing
model. Assumptions used were as follows: Fair value of the
underlying stock of $9.00, risk-free interest rate of 4.70%;
contractual life of 5 years; dividend yield of 0%; and
volatility of 70%. The fair value of the warrants amounted to
$416,000. This warrant was cancelled on January 24, 2007
and a new warrant, having an exercise price of $8.00 per share,
but otherwise having terms identical to the original warrant,
was issued to the director. The adjusted fair value of the
warrant amounted to $350,000. Following the guidance enumerated
in Issue 2 of
EITF 96-18,
the Company is amortizing the fair value of the warrants over
the two year term of the consulting agreement which is
consistent with its treatment of similar cash transactions. For
the year ended March 31, 2007, the amortized fair value of
the warrants amounted to $70,000 and was recorded as selling,
general and administrative expense in the accompanying
consolidated statements of operations.
On December 22, 2006, the Company issued a warrant to
purchase 50,000 shares of the Company’s common stock
at an exercise price of $3.00 per share in connection with a
settlement agreement with a former director and chief operating
officer. The warrants were non-forfeitable at date of issuance.
The warrants were valued using the Black-Scholes option pricing
model. Assumptions used were as follows: Fair value of the
underlying stock $9.00; risk-free interest rate 4.70%;
contractual life of 5 years; dividend yield of 0%; and
volatility of 70%. The fair value of the warrants amounted to
$365,000 and was recorded as selling, general and administrative
expense in the accompanying consolidated statement of operations
for the year ended March 31, 2007.
On June 1, 2006, the Company issued 3,750 shares of
common stock to a consultant in exchange for services provided.
The fair value of the underlying stock was valued at $11.28 per
share. The shares were fully vested and were non-forfeitable
when issued with no future performance obligation by the
consultant. The aggregate fair value of the shares, which
amounted to $43,000, was recorded as a selling, general and
administrative expense in the accompanying consolidated
statement of operations for the year March 31, 2007.
The Company accounted for its issuance of stock-based
compensation to non-employees for services using the
measurements date guidelines enumerated in SFAS 123(R) and
EITF 96-18.
Accordingly, the value of any awards that were vested and non
forfeitable at their date of issuance were measured based on the
fair value of the equity instruments at the date of issuance.
The non-vested portion of awards that are subject to the future
performance of the counterparty are adjusted at each reporting
date to their fair values based upon the then current market
value of the Company’s stock and other assumptions that
management believes are reasonable.
Valuation
of Common Stock
For the year ended March 31, 2004, all stock options that
the Company granted to employees and non-employees under its
1999, 2000 and 2003 Stock Option Plans were recorded at their
cash settlement value due to a compliance matter for which the
statute of limitations has expired. In July 2005, the Company
engaged Valuation
90
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Research Corporation, an outside valuation specialist to
determine the fair value of its common stock. The fair value of
the Company’s common stock, based on this valuation study,
was determined to be $10.16 per share. Accordingly, the fair
value of the Company’s common stock underlying all equity
transactions completed during the years ended March 31,
2004, 2005 and 2006 (other than options granted under the 1999,
2000 and 2003 Stock Option Plans) was based on the results of
the valuation study. The results were adjusted to the date of
grant based on an analysis performed by management. The results
were assessed for reasonableness by comparing such amounts to
concurrent sales of other equity instruments to unrelated
parties for cash and intervening events reflected in the price
of the Company’s stock.
In June 2006, the Company engaged Valuation Research
Corporation, an independent valuation specialist, to determine
the fair value of its common stock. The fair value of the
Company’s common stock, based on this valuation study, was
determined to be $11.28 per share. The fair value of the
Company’s common stock underlying common equity
transactions completed during the year ended March 31, 2007
was based on the valuation study, the Company’s estimate of
the mid-point of its proposed IPO price range, which was
determined in November 2006 to be $13.00 (subsequently reduced
in January 2007 to mid-point of $9.00) and a negotiated exercise
price of $18.00 per share for warrants issued to the placement
agent for the Series C convertible preferred stock offering.
|
|
|
NOTE 14 —
|
Stock-Based
Compensation
|
Reverse
Stock Split
On December 15, 2006, the Company effected an equity
restructuring through a
1-for-4
reverse stock split of its common stock. The Company split
adjusted both the exercise price and number of shares underlying
its outstanding employee stock options in accordance with stock
plan equity restructuring provisions, which include adjustments
for stock splits, contained in the Company’s stock option
plans. The Company applied the guidance specified in
paragraph 54 and the related implementation guidance
included in Appendix A of SFAS 123(R) to evaluate
whether the equity restructuring and modification of awards
resulted in an increase in the fair value of such awards and
whether additional compensation cost should be recognized. In
accordance with SFAS 123(R) awards that are modified in
equity restructurings pursuant to existing anti-dilution
provisions generally do not result in the recognition of
additional compensation cost. The Company evaluated the effect
of the reverse-split on the fair value of existing stock options
before and after the equity restructuring in accordance with the
equity restructuring guidelines. As a result, the Company
determined that it is not required to record additional
stock-based compensation cost.
1999,
2000, 2003 and 2004 Stock Option Plans
The 1999, 2000, 2003 and 2004 Stock Option Plans became
effective May 1999, June 2000, July 2003 and July 2004,
respectively. The Plans provide for grants of both incentive
stock options (ISO’s) and non-qualified stock options
(NSO’s) to employees, consultants and directors.
In accordance with the Plans, stated exercise price shall not be
less than 100% and 85% of the estimated fair market value of the
Company’s common stock on the date of grant for ISO’s
and NSO’s, respectively, as determined by the Board of
Directors at the date of grant. With respect to any 10%
shareholder, the exercise price of an ISO or NSO was not to
exceed 110% of the estimated fair market value per share on the
date of grant.
Options issued under the Plans generally have a ten-year term
and generally became exercisable over a five-year period.
As of June 29, 2006, the compensation committee of the
Company’s Board of Directors resolved that it would not
approve any further grants under its 1999, 2000, and 2003 Plans.
In connection with the reincorporation in Delaware, no future
options will be granted from the 2004 Plan.
91
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
2006
Stock Plan
On November 7, 2006, the Board authorized and reserved
1,250,000 shares for issuance of options that may be
granted under the Company’s 2006 Stock Incentive Plan
(“the 2006 Plan”), which was previously adopted by the
Board of Directors in August 2006. On December 14, 2006 the
stockholders approved the Company’s 2006 Plan which became
effective at the close of the Company’s initial public
offering.
The 2006 Plan provides for the granting of incentive stock
options to employees and the granting of nonstatutory stock
options to employees, non-employee directors, advisors, and
consultants. The 2006 Plan also provides for grants of
restricted stock, stock appreciation rights and stock units
awards to employees, non-employee directors, advisors and
consultants.
In accordance with the 2006 Plan, the stated exercise price
shall not be less than 100% and 85% of the estimated fair market
value of common stock on the date of grant for ISO’s and
NSO’s, respectively, as determined by the Board of
Directors at the date of grant. With respect to any 10%
stockholder, the exercise price of an ISO or NSO shall not be
less than 110% of the estimated fair market value per share on
the date of grant.
Options issued under the 2006 Plan generally have a ten-year
term and generally become exercisable over a five-year period.
Shares subject to awards that expire unexercised or are
forfeited or terminated will again become available for issuance
under the 2006 Plan. No participant in the 2006 Plan can receive
option grants, restricted shares, stock appreciation rights or
stock units for more than 750,000 shares in the aggregate
in any calendar year.
As of March 31, 2007, 1,115,000 shares remain
authorized for issuance under the 2006 Plan and 135,000 options
are issued and outstanding.
Options
Granted Outside of Plans
In May 2004, the Company granted an option to purchase
300,000 shares of the Company’s common stock with an
exercise price of $0.16 per share to the Chief Executive Officer
of the Company. The fair value of the underlying common stock at
the date of grant was $5.96 per share. The options were fully
exercisable on the date of grant. Stock-based compensation
expense related to these options amounted to $1,740,000 and was
recorded in selling, general and administrative expense in the
consolidated statement of operations for the year ended
March 31, 2005.
Options
Subject to Repurchase
During the period from May 1999 to December 2003, the Company
granted an aggregate of 1,827,405 stock options to various
employees and non-employees under its 1999, 2000, and 2003
Plans. Subsequent to making such grants, the Company determined
that such grants may not have been exempt from registration or
qualification rights under the provisions of applicable state
securities laws. A failure to comply with applicable state
securities laws may give rise to claims of optionees against the
Company for the repurchase of their unexercised options at an
amount determined by a formula specified by state securities law
regulators, plus legal interest, or rescission of the purchase
of the shares of common stock issued upon exercise of the
options at an amount equal to the exercise price of the options,
plus interest from the date of exercise. The repurchase and
rescission rights held by the Company’s security holders,
if any, are subject to applicable statute of limitations
prescribed by state law. In California, the statute of
limitation is two years. During the period from May 2001 to
December 2005 the statute of limitations would have lapsed for
bringing claims against the Company related to options granted
during the period from May 2001 to December 2005 subject to
California law.
The Company accounted for the repurchase and rescission rights
in accordance with APB 25 paragraph 25 and SFAS 123
paragraph 25, both of which are titled “Awards That
Call for Settlement in Cash”. These standards require
entities to record stock-based compensation awards as liability
instruments when the optionee has the
92
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
ability to compel the entity to settle the award by transferring
cash or other assets. In addition, other accounting literature
(including literature relating to accounting for derivative
financial instruments) requires liability classification when a
net cash settlement is in the holder’s control. The Company
believes that if the holders of these awards possess a free
standing right to require cash settlement that liability
classification of these awards is required under APB 25 and
SFAS 123 (the standards applicable at the time of grant)
and that such treatment is consistent with the principles of
other literature relating to the classification of financial
instruments. Accordingly, these awards were classified as
liability instruments for their estimated cash settlement
amounts. The Company reclassified the liability instruments to
permanent equity at the time the statute of limitations lapsed
and the holder could no longer control settlement of the award
in cash.
During the year ended March 31, 2006 and 2005 the Company
included in the accompanying consolidated statements of
operations stock compensation expense of $6,000 and $22,000,
respectively.
A summary of activity under option Plans for the years ended
March 31, 2007, 2006 and 2005 is presented below (in
thousands, except per share data):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Aggregate
|
|
|
|
|
Number of
|
|
|
Weighted-Average
|
|
|
Weighted-Average
|
|
|
Intrinsic
|
|
|
|
|
Shares
|
|
|
Exercise Price
|
|
|
Contractual Term
|
|
|
Value
|
|
|
|
|
Outstanding at March 31, 2004
|
|
|
1,535
|
|
|
$
|
1.35
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
613
|
|
|
|
1.61
|
|
|
|
|
|
|
|
|
|
|
Options forfeited or expired
|
|
|
(508
|
)
|
|
|
1.30
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at March 31, 2005
|
|
|
1,640
|
|
|
|
1.46
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
787
|
|
|
|
9.20
|
|
|
|
|
|
|
|
|
|
|
Options exercised
|
|
|
(292
|
)
|
|
|
1.02
|
|
|
|
|
|
|
|
|
|
|
Options forfeited or expired
|
|
|
(166
|
)
|
|
|
6.17
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at March 31, 2006
|
|
|
1,969
|
|
|
|
4.22
|
|
|
|
|
|
|
|
|
|
|
Options granted
|
|
|
380
|
|
|
|
9.20
|
|
|
|
|
|
|
|
|
|
|
Options forfeited or expired
|
|
|
(329
|
)
|
|
|
5.76
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at March 31, 2007
|
|
|
2,020
|
|
|
$
|
4.91
|
|
|
|
6.72
|
|
|
$
|
5,566
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at March 31, 2007
|
|
|
1,317
|
|
|
$
|
2.68
|
|
|
|
5.83
|
|
|
$
|
5,127
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options available for grant as of
March 31, 2007
|
|
|
1,115
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The aggregate intrinsic value is calculated as the difference
between the exercise price of the underlying stock options and
the fair value of the Company’s common stock at the time
($5.95) for stock options that are in-the-money as of
March 31, 2007.
Stock-Based
Compensation Before Adoption of
SFAS No. 123(R)
Prior to April 1, 2006, the Company accounted for
stock-based employee compensation arrangements in accordance
with the provisions of APB No. 25, “Accounting for
Stock Issued to Employees,” and its related interpretations
and applied the disclosure requirements of
SFAS No. 148, “Accounting for Stock-Based
Compensation-Transition and Disclosure, an amendment of FASB
Statement No. 123.” The Company used the minimum value
method to measure the fair value of awards issued prior to
April 1, 2006 with respect to its application of the
disclosure requirements under SFAS 123.
93
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The following table illustrates the effect on net loss as if the
Company had applied the fair value recognition provisions of
SFAS 123 to stock-based compensation arrangements (in
thousands, except per share data):
| |
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2006
|
|
|
2005
|
|
|
|
|
Net loss available to common
stockholders, as reported
|
|
$
|
(23,220
|
)
|
|
$
|
(16,530
|
)
|
|
Add: Total stock-based employee
compensation expenses included in net loss
|
|
|
279
|
|
|
|
2,297
|
|
|
Deduct: Total stock-based employee
compensation determined under the fair-value based method for
all awards
|
|
|
(503
|
)
|
|
|
(2,448
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net loss available to common
stockholders, pro forma
|
|
$
|
(23,444
|
)
|
|
$
|
(16,681
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share, basic
and diluted:
|
|
|
|
|
|
|
|
|
|
As reported
|
|
$
|
(5.60
|
)
|
|
$
|
(4.22
|
)
|
|
Pro forma
|
|
$
|
(5.65
|
)
|
|
$
|
(4.26
|
)
|
In accordance with the provisions of SFAS No. 123, the
fair value of each employee option granted in reporting periods
prior to the adoption of SFAS 123(R) was estimated on the
date of grant using the minimum value method with the following
weighted-average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2006
|
|
|
2005
|
|
|
|
|
Estimated life
|
|
|
6 yrs
|
|
|
|
6 yrs
|
|
|
Risk-free interest rate
|
|
|
4.27
|
%
|
|
|
3.95
|
%
|
|
Dividend yield
|
|
|
0.00
|
%
|
|
|
0.00
|
%
|
The weighted-average estimated minimum values of options granted
were $3.12 and $5.00 for the years ended March 31, 2006 and
2005, respectively.
Stock-Based
Compensation After Adoption of SFAS 123(R)
Effective April 1, 2006, the Company adopted
SFAS No. 123(R), Share-Based Payment, using the
prospective transition method, which requires the measurement
and recognition of compensation expense for all share-based
payment awards granted, modified and settled to the
Company’s employees and directors after April 1, 2006.
The Company’s consolidated financial statements as of and
for the year ended March 31, 2007 reflect the impact of
SFAS No. 123(R). In accordance with the prospective
transition method, the Company’s consolidated financial
statements for prior periods have not been restated to reflect,
and do not include, the impact of SFAS No. 123(R).
The effect of the change of recording stock-based compensation
expense from the original provisions of APB No. 25 to the
provisions of SFAS No. 123(R)for the year ended
March 31, 2007 is as follows (in thousands, except per
share amounts):
| |
|
|
|
|
|
|
|
Impact from
|
|
|
|
|
SFAS No. 123(R)
|
|
|
|
|
Provisions for
|
|
|
|
|
the Year Ended
|
|
|
|
|
March 31, 2007
|
|
|
|
|
Cost of revenues service
|
|
$
|
3
|
|
|
Selling, general and administrative
|
|
|
812
|
|
|
|
|
|
|
|
|
Total stock-based compensation
|
|
$
|
815
|
|
|
|
|
|
|
|
|
Effect on basic and diluted net
loss per common share
|
|
$
|
0.15
|
|
|
|
|
|
|
|
94
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
No income tax benefit has been recognized relating to
stock-based compensation expense and no tax benefits have been
realized from exercised stock options. The implementation of
SFAS No. 123(R) did not have an impact on cash flows
from financing activities during the year ended March 31,
2007.
The Company estimated the fair value of employee stock options
using the Black-Scholes option pricing model. The fair value of
employee stock options is being amortized on a straight-line
basis over the requisite service period of the awards. The fair
value of employee stock options was estimated using the
following weighted-average assumptions for the year ended
March 31, 2007:
| |
|
|
|
|
|
|
|
Year Ended
|
|
|
|
|
March 31, 2007
|
|
|
|
|
Expected Term
|
|
|
3.95
|
yrs
|
|
Risk-free interest rate
|
|
|
4.60
|
%
|
|
Dividend yield
|
|
|
0.00
|
%
|
|
Volatility
|
|
|
70.0
|
%
|
The expected term of stock options represents the average period
the stock options are expected to remain outstanding and is
based on the expected term calculated using the approach
prescribed by SAB 107 for “plain vanilla”
options. The Company used this approach as it did not have
sufficient historical information to develop reasonable
expectations about future exercise patterns and post-vesting
employment termination behavior. The expected stock price
volatility for the Company’s stock options for the year
ended March 31, 2007 was determined by examining the
historical volatilities for industry peers and using an average
of the historical volatilities of the Company’s industry
peers as the Company did not have any trading history for the
Company’s common stock. The Company will continue to
analyze the historical stock price volatility and expected term
assumption as more historical data for the Company’s common
stock becomes available. The risk-free interest rate assumption
is based on the U.S. Treasury instruments whose term was
consistent with the expected term of the Company’s stock
options. The expected dividend assumption is based on the
Company’s history and expectation of dividend payouts.
In addition, SFAS No. 123(R) requires forfeitures to
be estimated at the time of grant and revised, if necessary, in
subsequent periods if actual forfeitures differ from those
estimates. Forfeitures were estimated based on historical
experience. Prior to the adoption of SFAS No. 123(R),
the Company accounted for forfeitures as they occurred.
At March 31, 2007, there was $343,000 of unrecognized
compensation cost related to options that the Company accounted
for under APB 25 through March 31, 2006. These costs are
expected to be recognized over a weighted average amortization
period of 3.31 years.
During the year ended March 31, 2007, the Company granted
380,124 stock options to employees with a weighted-average grant
date fair value of $9.20 per share. At March 31, 2007,
there was unrecognized compensation costs of $1,156,000 related
to these stock options. The cost is expected to be recognized
over a weighted-average amortization period of 3.32 years.
During the year ended March 31, 2007, the Company modified
stock options granted to employees and non-employees under share
based arrangements in connection with the reverse-stock split
equity restructuring. As described previously, the Company was
not required to record any additional compensation in connection
with the reverse-stock split. In addition, the Company modified
an option grant to a Board Member, Robert Burlingame. In
accordance with his agreement with the Company, the exercise
price of the 75,000 options granted would be equal to the IPO
price of $8.00. On January 25, 2007, the Company cancelled
and regranted the options at the price of $8.00. The Company
treated the cancellation and regrant as a modification to the
original grant and recorded incremental compensation cost of
$22,000.
The Company did not capitalize any cost associated with
stock-based compensation.
The Company issues new shares of common stock upon exercise of
stock options.
95
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Non-Employee
Options
The Company believes that the fair value of the stock options
issued to non-employees is more reliably measurable than the
fair value of the services received. The fair value of the stock
options granted was calculated using the Black-Scholes
option-pricing model as prescribed by SFAS No. 123
using the following weighted-average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
Estimated life
|
|
|
8.83 yrs
|
|
|
|
8.67 yrs
|
|
|
|
9.06 yrs
|
|
|
Risk-free interest rate
|
|
|
4.29
|
%
|
|
|
4.27
|
%
|
|
|
4.50
|
%
|
|
Dividend yield
|
|
|
0.00
|
%
|
|
|
0.00
|
%
|
|
|
0.00
|
%
|
|
Volatility
|
|
|
70
|
%
|
|
|
70
|
%
|
|
|
70
|
%
|
The stock-based compensation expense will fluctuate as the fair
market value of the common stock fluctuates. In connection with
stock options granted to non-employees, the Company recorded
$11,000, $32,000 and $30,000 of stock-based compensation expense
in the years ended March 31, 2007, 2006 and 2005,
respectively.
The Company has the following net deferred tax assets (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
Deferred tax assets:
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards
|
|
$
|
23,248
|
|
|
$
|
17,290
|
|
|
Tax credit carryforwards
|
|
|
420
|
|
|
|
212
|
|
|
Stock-based compensation
|
|
|
1,664
|
|
|
|
1,070
|
|
|
Reserves and accruals
|
|
|
1,013
|
|
|
|
186
|
|
|
Other deferred tax assets
|
|
|
173
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax assets
|
|
$
|
26,518
|
|
|
$
|
18,758
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred tax liabilities:
|
|
|
|
|
|
|
|
|
|
Basis difference in assets
|
|
|
(20
|
)
|
|
|
(78
|
)
|
|
State taxes
|
|
|
(1,766
|
)
|
|
|
(897
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total deferred tax liabilities
|
|
|
(1,786
|
)
|
|
|
(975
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset
|
|
|
24,732
|
|
|
|
17,783
|
|
|
Valuation allowance
|
|
|
(24,732
|
)
|
|
|
(17,783
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax asset
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The Company’s recorded income tax benefit, net of the
change in the valuation allowance, for each of the periods
presented is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
Income tax benefit
|
|
$
|
6,949
|
|
|
$
|
8,107
|
|
|
$
|
6,019
|
|
|
Change in valuation allowance
|
|
|
(6,949
|
)
|
|
|
(8,107
|
)
|
|
|
(6,019
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income tax benefit
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
96
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
A reconciliation of the statutory federal income tax rate to the
Company’s effective tax rate is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2005
|
|
|
|
|
Expected statutory rate
|
|
|
(34.0
|
)%
|
|
|
(34.0
|
)%
|
|
|
(34.0
|
)%
|
|
State income taxes, net of federal
benefit
|
|
|
(5.8
|
)%
|
|
|
(3.3
|
)%
|
|
|
(3.8
|
)%
|
|
Research and Development Credit
|
|
|
(0.7
|
)%
|
|
|
—
|
|
|
|
—
|
|
|
Foreign earnings taxed at
different rates
|
|
|
2.6
|
%
|
|
|
1.8
|
%
|
|
|
1.0
|
%
|
|
Japan loss not benefited
|
|
|
0.2
|
%
|
|
|
—
|
|
|
|
—
|
|
|
Effect of permanent differences
|
|
|
2.6
|
%
|
|
|
0.3
|
%
|
|
|
0.3
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(35.1
|
)%
|
|
|
(35.2
|
)%
|
|
|
(36.5
|
)%
|
|
Change in valuation allowance
|
|
|
35.1
|
%
|
|
|
35.2
|
%
|
|
|
36.5
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Totals
|
|
|
0.0
|
%
|
|
|
0.0
|
%
|
|
|
0.0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At March 31, 2007, the Company had net operating loss
carryforwards for federal, state and foreign income tax purposes
of approximately $40,093,000, $53,010,000 and $18,238,000,
respectively. The carryforwards expire at various times through
March 31, 2020. The Company also had, at March 31,
2007, federal and state research and development credit
carryforwards of approximately $206,000 and $214,000,
respectively. The federal credits expire beginning in 2026 and
the state credits do not expire.
The Company experienced substantial ownership changes in
connection with financing transactions it completed during the
year ended March 31, 2006. Accordingly, the Company’s
utilization of its net operating loss and tax credit
carryforwards against taxable income in future periods, if any,
is subject to substantial limitations under the Change in
Ownership rules of Section 382 of the Internal Revenue
Code. The Company, after considering all available evidence,
fully reserved for these and its other deferred tax assets since
it is more likely than not such benefits will not be realized in
future periods. The Company has incurred losses for both
financial reporting and income tax purposes for the year ended
March 31, 2007. Accordingly, the Company is continuing to
fully reserve for its deferred tax assets. The Company will
continue to evaluate its deferred tax assets to determine
whether any changes in circumstances could affect the
realization of their future benefit. If it is determined in
future periods that portions of the Company’s deferred
income tax assets satisfy the realization standard of
SFAS No. 109, the valuation allowance will be reduced
accordingly.
|
|
|
NOTE 16 —
|
Employee
Benefit Plan
|
In 2002, the Company established a program to contribute and
administer individual retirement accounts for regular full time
employees. Under the plan the Company matches employee
contributions to the plan up to 3% of the employee’s
salary. The Company contributed $66,000, $53,000 and $63,000 to
the program for the years ended March 31, 2007, 2006 and
2005, respectively.
|
|
|
NOTE 17 —
|
Segment
and Geographic Information
|
In accordance with SFAS No. 131, “Disclosures
About Segments of an Enterprise and Related Information”
(“SFAS 131”), operating segments are identified
as components of an enterprise for which separate and discreet
financial information is available and is used by the chief
operating decision maker, or decision-making group, in making
decisions on how to allocate resources and assess performance.
The Company’s chief decision-makers, as defined by
SFAS 131, are the Chief Executive Officer and his direct
reports.
The Company’s chief decision-makers review financial
information presented on a consolidated basis, accompanied by
disaggregated information about revenue and operating profit by
operating unit. This information is used for purposes of
allocating resources and evaluating financial performance.
97
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The accounting policies of the segments are the same as those
described in the “Summary of Significant Accounting
Policies.” Segment data includes segment revenue, segment
operating profitability, and total assets by segment. Shared
corporate operating expenses are reported in the
U.S. segment.
The Company is organized primarily on the basis of operating
units which are segregated by geography. Oculus Japan is
insignificant with respect to the Company’s consolidated
operating results for the year ended March 31, 2007 and
therefore has been included in the U.S. segment.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S.
|
|
|
Europe
|
|
|
Mexico
|
|
|
Total
|
|
|
|
|
Year Ended March 31,
2007:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenues
|
|
$
|
140
|
|
|
$
|
1,026
|
|
|
$
|
2,513
|
|
|
$
|
3,679
|
|
|
Service revenues
|
|
|
864
|
|
|
|
—
|
|
|
|
—
|
|
|
|
864
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
1,004
|
|
|
|
1,026
|
|
|
|
2,513
|
|
|
|
4,543
|
|
|
Depreciation expense
|
|
|
377
|
|
|
|
203
|
|
|
|
92
|
|
|
|
672
|
|
|
Operating loss
|
|
|
(13,066
|
)
|
|
|
(2,905
|
)
|
|
|
(3,513
|
)
|
|
|
(19,484
|
)
|
|
Interest expense
|
|
|
(956
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(956
|
)
|
|
Interest income
|
|
|
312
|
|
|
|
—
|
|
|
|
—
|
|
|
|
312
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S.
|
|
|
Europe
|
|
|
Mexico
|
|
|
Total
|
|
|
|
|
Year Ended March 31,
2006:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenues
|
|
$
|
109
|
|
|
$
|
69
|
|
|
$
|
1,788
|
|
|
$
|
1,966
|
|
|
Service revenues
|
|
|
618
|
|
|
|
—
|
|
|
|
—
|
|
|
|
618
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
727
|
|
|
|
69
|
|
|
|
1,788
|
|
|
|
2,584
|
|
|
Depreciation expense
|
|
|
463
|
|
|
|
96
|
|
|
|
92
|
|
|
|
651
|
|
|
Operating loss
|
|
|
(12,621
|
)
|
|
|
(2,685
|
)
|
|
|
(5,545
|
)
|
|
|
(20,851
|
)
|
|
Interest expense
|
|
|
(172
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(172
|
)
|
|
Interest income
|
|
|
282
|
|
|
|
—
|
|
|
|
—
|
|
|
|
282
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S.
|
|
|
Europe
|
|
|
Mexico
|
|
|
Total
|
|
|
|
|
Year Ended March 31,
2005:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product revenues
|
|
$
|
4
|
|
|
$
|
35
|
|
|
$
|
434
|
|
|
$
|
473
|
|
|
Service revenues
|
|
|
883
|
|
|
|
—
|
|
|
|
—
|
|
|
|
883
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
887
|
|
|
|
35
|
|
|
|
434
|
|
|
|
1,356
|
|
|
Depreciation expense
|
|
|
368
|
|
|
|
49
|
|
|
|
17
|
|
|
|
434
|
|
|
Operating loss
|
|
|
(12,242
|
)
|
|
|
(1,529
|
)
|
|
|
(2,541
|
)
|
|
|
(16,312
|
)
|
|
Interest expense
|
|
|
(372
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(372
|
)
|
|
Interest income
|
|
|
8
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8
|
|
For the year ended March 31, 2007, sales to a customer in
India were $604,000. These sales are reported as part of the
Europe segment.
98
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The following table shows property and equipment balances by
segment (in thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
U.S.
|
|
$
|
904
|
|
|
$
|
930
|
|
|
Europe
|
|
|
901
|
|
|
|
639
|
|
|
Mexico
|
|
|
402
|
|
|
|
371
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
2,207
|
|
|
$
|
1,940
|
|
|
|
|
|
|
|
|
|
|
|
The following table shows total asset balances by segment (in
thousands):
| |
|
|
|
|
|
|
|
|
|
|
|
March 31,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
|
|
U.S.
|
|
$
|
23,437
|
|
|
$
|
8,977
|
|
|
Europe
|
|
|
1,367
|
|
|
|
1,652
|
|
|
Mexico
|
|
|
2,146
|
|
|
|
2,060
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
26,950
|
|
|
$
|
12,689
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NOTE 18 —
|
Discontinued
Operations
|
On June 16, 2005, the Company entered into a series of
agreements with Quimica Pasteur, or QP, a Mexico-based company
engaged in the business of distributing pharmaceutical products
to hospitals and health care entities owned or operated by the
Mexican Ministry of Health. These agreements provided, among
other things, for QP to act as the Company’s exclusive
distributor of Microcyn to the Mexican Ministry of Health for a
period of three years. The Company was granted an option to
acquire the remaining 99.75% directly from its principals in
exchange for 600,000 shares of common stock, contingent
upon QP’s attainment of certain financial milestones. The
Company’s distribution and related agreements were
cancelable by the Company on thirty days’ notice without
cause and included certain provisions to hold the Company
harmless from debts incurred by QP outside the scope of the
distribution and related agreements. The Company terminated
these agreements on March 26, 2006.
Due to its liquidity circumstances, QP was unable to sustain
operations without the Company’s subordinated financial and
management support. Accordingly, QP was deemed to be a variable
interest entity in accordance with FIN 46(R) and its
results were consolidated with the Company’s consolidated
financial statements for the period of June 16, 2005
through March 26, 2006, the effective termination date of
the distribution and related agreements.
In accordance with SFAS 144, the Company has reported
QP’s results for the period of June 16, 2005 through
March 26, 2006 as discontinued operations because the
operations and cash flows of QP have been eliminated from the
Company’s ongoing operations as a result of having
terminated these agreements. The Company no longer has any
continuing involvement with QP as of the date in which the
agreements were terminated. Amounts associated with the
Company’s loss upon the termination of its agreements with
QP, which consists of funds advanced by the Company for working
capital, are presented separately from QP’s operating
results.
Subsequent to having entered into the agreements with QP, the
Company became aware of an alleged tax avoidance scheme
involving the principals of QP. The audit committee of the
Company’s Board of Directors engaged an independent
counsel, as well as tax counsel in Mexico to investigate this
matter. The audit committee of the Board of Directors was
advised that QP’s principals could be liable for up to
$7,000,000 of unpaid taxes; however, the Company is unlikely to
have any loss exposure with respect to this matter because the
alleged tax omission occurred prior to the Company’s
involvement with QP. The Company has not received any
communications to date from Mexican tax authorities with respect
to this matter.
99
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Based on an opinion of Mexico counsel, the Company management
and the audit committee of the Board of Directors do not believe
that the Company is likely to experience any loss with respect
to this matter. However, there can be no assurance that the
Mexican tax authorities will not pursue this matter and, if
pursued, that it would not result in a material loss to the
Company.
|
|
|
NOTE 19 —
|
Selected
Quarterly Financial Data (unaudited)
|
The Company believes that the following information reflects all
adjustments, consisting of only normal recurring adjustments,
necessary for a fair presentation of the information for the
periods presented. The operating results for any quarter are not
necessarily indicative of results for any future periods.
The following table contains selected unaudited statements of
operations information for each of the quarters for the years
ended March 31, 2007 and 2006 (in thousands, except per
share data):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
|
March 31,
|
|
|
December 31,
|
|
|
September 30,
|
|
|
June 30,
|
|
|
|
|
2007
|
|
|
2006
|
|
|
2006
|
|
|
2006
|
|
|
|
|
Revenue
|
|
$
|
1,162
|
|
|
$
|
1,052
|
|
|
$
|
1,252
|
|
|
$
|
1,078
|
|
|
Gross profit
|
|
|
388
|
|
|
|
292
|
|
|
|
492
|
|
|
|
373
|
|
|
Net loss available to common
stockholders
|
|
|
(6,332
|
)
|
|
|
(4,948
|
)
|
|
|
(4,498
|
)
|
|
|
(4,418
|
)
|
|
Basic and diluted net loss per
common share
|
|
$
|
(0.69
|
)
|
|
$
|
(1.17
|
)
|
|
$
|
(1.06
|
)
|
|
$
|
(1.05
|
)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quarter Ended
|
|
|
|
|
March 31,
|
|
|
December 31,
|
|
|
September 30,
|
|
|
June 30,
|
|
|
|
|
2006
|
|
|
2005
|
|
|
2005
|
|
|
2005
|
|
|
|
|
Revenue
|
|
$
|
922
|
|
|
$
|
581
|
|
|
$
|
676
|
|
|
$
|
406
|
|
|
Gross (loss)
|
|
|
(303
|
)
|
|
|
(1,249
|
)
|
|
|
(432
|
)
|
|
|
(333
|
)
|
|
Net loss available to common
stockholders
|
|
|
(7,396
|
)
|
|
|
(6,080
|
)
|
|
|
(5,602
|
)
|
|
|
(4,142
|
)
|
|
Basic and diluted net loss per
common share
|
|
$
|
(1.75
|
)
|
|
$
|
(1.45
|
)
|
|
$
|
(1.33
|
)
|
|
$
|
(1.04
|
)
|
Per common share amounts for the quarters and full years have
been calculated separately. Accordingly, quarterly amounts do
not add to the annual amount because of differences in the
weighted average common shares outstanding during each period
principally due to the effect of the Company’s issuing
shares of its common stock during the year.
Diluted and basic net loss per common share are identical since
common equivalent shares are excluded from the calculation, as
their effect is anti-dilutive.
|
|
|
NOTE 20 —
|
Subsequent
Events
|
Increase
in Number of Shares Authorized in 2006 Plan
As provided under the 2006 Plan, the aggregate number of shares
authorized for issuance as awards under the 2006 Plan
automatically increased on April 1, 2007 by
592,220 shares (which number constitutes 5% of the
outstanding shares on the last day of the fiscal year ended
March 31, 2007). Total shares authorized for issuance
subsequent to the increase is 1,842,220.
Board
Compensation
On April 26, 2007, the Board of Directors of the Company
adopted a Board Compensation Package (the “Compensation
Package”) to provide members of the Board and its
committees with regular compensation. The Compensation Package
provides for cash payments of $25,000 in two equal installments
to each of the non-employee members of the Board of Directors.
Directors who are members (but not the chairman) of the Audit
100
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Committee will receive an additional $5,000 per year. Directors
who are members (but not the chairman) of the Compensation
Committee will receive an additional $2,000 per year. The chair
person of the Board of Directors will receive $15,000 annually,
the Lead Director (if different from the chair person) will
receive $10,000 annually, the chairperson of the audit committee
will receive $10,000 annually, and the chair person of each
other committee will receive $5,000 annually.
The Compensation Committee also recommended to the Board the
amendment and restatement of the Company’s 2006 Stock
Incentive Plan to include provisions concerning automatic grants
to non-employee directors, and the Board adopted such changes,
subject to approval of the stockholders. The Compensation
Package provides for the grant of options to each non-employee
director under the restated Stock Incentive Plan, subject to
stockholder approval. If approved by the stockholders, each new
director would receive an initial option grant to purchase
50,000 shares of the Company’s Common Stock, which
will vest over three years, and each non-employee director will
receive an annual grant of an option to purchase
15,000 shares of the Company’s Common Stock, which
will vest monthly over a period of one year.
Stock
Unit Grant to Officer
On October 1, 2005, the Board authorized the grant to one
of the Company’s officers at the closing of its initial
public offering of an option under the 2004 Stock Option Plan to
purchase 60,000 shares of the Company’s common stock
at an exercise price of $3.00 per share. Due to the Board’s
subsequent decision not to grant additional options under the
2004 Stock Option Plan, the adoption by the Board and approval
by the stockholders of the Company’s 2006 Stock Incentive
Plan, and certain regulatory developments, on April 26,
2007, in lieu of the award of an option, the Compensation
Committee authorized the award to the officer of 60,000 stock
units. Each stock unit represents the right to receive a share
of the Company’s common stock, in consideration of past
services rendered and the payment by the officer of $3.00 per
share, upon the settlement of the stock unit on a fixed date in
the future. Half of the stock units, representing
30,000 shares, will be settled on January 15, 2009 and
the remaining 30,000 will be settled on January 15, 2010.
Acceleration
of Vesting
On April 26, 2007 the Company terminated certain advisory
consulting contracts and made all unvested warrants issued to
the consultants available for immediate exercise. In addition,
the Company extended the exercise period through April 13, 2009.
Bonus
Plan
On June 15, 2007, the compensation committee of the board
of directors approved a bonus plan for fiscal year 2008 (the
“Bonus Plan”), under which all employees, including
executive officers, are eligible for bonus awards if the Company
attains specified targeted company goals and the individual
obtains his or her individual performance goals. Bonuses, if
awarded, are payable in cash or, at the determination of the
Compensation Committee no later than April 7, 2008 as
necessary to preserve our cash reserves, in part or in whole in
grants of stock options under the 2006 Stock Incentive Plan. Any
stock options will be valued by reference to the Black Sholes
option pricing calculation at the fair market value of the
Company’s common stock on June 7, 2008. However, the
compensation committee may not grant options exceeding the
number of shares authorized under the 2006 Stock Incentive Plan
less the number of shares the Company is contractually obligated
to grant in the next year.
The amount of the awards under the plan will depend on the
company achieving specified targeted revenue, expense, cash
position and operational goals, and on the employee achieving
his or her individual goals. The determination of whether the
financial and performance goals have been met will be in the
sole discretion of the compensation committee. In addition, the
compensation committee will establish and take into
consideration additional yearly operational goals, including
milestones for clinical trials, research and development,
quality assurance and corporate development, in determining
whether any adjustment to the target bonus should be
101
OCULUS
INNOVATIVE SCIENCES, INC. AND SUBSIDIARIES
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
adjusted upwards. Stretch bonuses constitute an upper limit on
the bonus potential for employees. Under the BonusPlan, no bonus
is paid unless the Company meets specified minimum revenue,
operating expense and cash position targets for fiscal 2008, and
no bonus will be paid unless an individual’s performance
goals are achieved. The award of any bonus, other than the
guaranteed cash bonuses in the amount of $50,000 each to two
executive officers, and the bonuses for fiscal year 2007
specified in the Bonus Plan, is at the sole discretion of the
compensation committee.
Upon achievement of minimum revenue, expense and cash position
targets and individual goals (the “Minimum Target”),
employees (other than executive officers) are eligible to earn a
threshold award of 10% of their individual base salary or
additional stock option grants, at the discretion of the
compensation committee. Upon achievement of 100% of revenue,
expense and cash position targets and certain operational
targets, and achievement of individual goals (the “Stretch
Target”), employees (other than executive officers) are
eligible to earn a bonus award of up to 35% of their individual
base salary. If the compensation committee determines to award
one or more bonuses, and if Target Milestones are exceeded, but
Stretch Milestones are not achieved, the compensation committee
may, at its discretion, award a bonus in an amount between the
Target Milestone bonus and Stretch Milestone bonus amount.
Upon achievement of Minimum Target goals and Stretch Target
goals, the chief executive officer will be eligible to earn a
bonus of 100% to 200% of his individual base salary; and the
chief operating officer, the chief financial officer and the
vice president corporate development and general counsel will be
eligible to earn a bonus of 50% to 150% of his respective
individual base salary. The vice president operations and
international sales is eligible to receive up to $100,000 in
bonus for fiscal year 2008, which will be paid quarterly, if
awarded, and up to an additional $50,000 cash bonus payable at
the end of the fiscal year, based on achievement of certain
revenue and net operating income goals. The Company hired a vice
president of regulatory and clinical trials who commenced his
employment on June 18, 2007, whose annual base salary is
$242,000. The compensation committee has authorized a guaranteed
cash bonus of $50,000 for fiscal year 2008 after the end of 2008.
The Bonus Plan also authorized the payment of a cash bonus in
the amount of $150,000 to the chief executive officer and a cash
bonus in the amount of $60,000 to the vice president corporate
development and general counsel which were paid on June 15,
2007. In addition, on June 15, 2007, the compensation committee
authorized the immediate grant of an option to purchase
150,000 shares of our common stock to the chief operating
officer, an option to purchase 25,000 of our common stock to the
vice president operations and international sales, and an option
to purchase 100,000 shares of our common stock to the vice
president corporate development and general counsel, in each
case based upon each employee’s and the Company’s
performance in fiscal year 2007. Each option will be granted
with an exercise price equal to the closing price of the
Company’s common stock on June 15, 2007.
102
|
|
|
ITEM 9.
|
Changes
in and disagreements with Accountants on Accounting and
Financials Disclosure
|
On April 12, 2006, the Audit Committee of our board of
directors approved the dismissal of PricewaterhouseCoopers LLP,
or PWC, as our independent registered public accounting firm and
subsequently appointed Marcum & Kliegman LLP, or
M&K, effective April 12, 2006. We did not consult with
M&K on any accounting or financial reporting matters prior
to M&K’s appointment.
We engaged PWC on June 14, 2005, to perform an audit of our
financial statements for our fiscal years ended March 31,
2003, 2004 and 2005. PWC did not issue a report on our financial
statements for the years ended March 31, 2004 or 2005, or
through April 12, 2006. For the years ended March 31,
2003, 2004 and 2005, and through April 12, 2006, there were
no disagreements with PWC on any matter of accounting principles
or practices, financial statement disclosure or auditing scope
or procedure, which disagreements, if not resolved to PWC’s
satisfaction, would have caused PWC to make reference thereto in
their report on the financial statements for such years if they
had delivered a report. In March 2006, and prior to its
dismissal, PWC advised our Audit Committee orally of the
following:
|
|
|
| |
•
|
the absence of financial accounting personnel with sufficient
skills and experience to effectively evaluate and determine the
appropriate accounting for non-routine
and/or
complex accounting transactions consistent with accounting
principles generally accepted in the United States, which
resulted in a number of material audit adjustments to the
financial statements during the course of audit procedures;
|
| |
| |
•
|
the failure to maintain effective controls to ensure the
identification of accounting issues related to and the proper
accounting for stock options with the right of rescission that
were granted under certain stock option plans that required
registration or qualification under federal and state securities
laws primarily due to insufficient oversight and lack of
personnel in the accounting and finance organization with the
appropriate level of accounting knowledge, experience and
training;
|
| |
| |
•
|
the failure to maintain an effective anti-fraud program designed
to detect and prevent fraudulent activities in QP;
|
| |
| |
•
|
the need to expand significantly the scope of the audit of QP to
assess the impact of identified fraudulent activities on the our
financial statements, in which regard PWC advised our audit
committee that the results of the fraud investigation may cause
PWC to be unwilling to be associated with our financial
statements;
|
| |
| |
•
|
the “tone at the top” set by our senior management
does not appear to encourage an attitude within our company that
controls are important and that established controls cannot be
circumvented;
|
| |
| |
•
|
we did not have the appropriate financial management and
reporting infrastructure in place to meet the demands that will
be placed upon us as a public company, including the
requirements of the Sarbanes-Oxley Act of 2002, and that we will
be unable to report our financial results accurately or in a
timely manner; and
|
| |
| |
•
|
significant control deficiencies, when considered in the
aggregate, constituted a material weakness over financial
reporting.
|
We have authorized PWC to respond fully to the inquiries of
M&K concerning the foregoing. We have taken the following
steps designed to address PWC’s concerns and to implement
the recommendations made by our special counsel to our audit
committee in connection with its investigation of QP:
|
|
|
| |
•
|
we have implemented a training program to continue to enhance
the knowledge and skills of our finance personnel on accounting
developments and the application of generally accepted
accounting principles to non-routine and complex transactions
commensurate with our financial reporting requirements;
|
| |
| |
•
|
we have implemented programs to enhance the knowledge and skills
of our employees in finance responsible for overseeing the
consolidation of financial results of any subsidiary, foreign or
domestic, to accumulate the necessary accounting, finance and
operational information to effectively analyze information
required for financial statement preparation and footnote
disclosures, including the identification of potential fraud;
|
103
|
|
|
| |
•
|
we have hired a director of finance and compliance with relevant
accounting, audit and compliance experience, skills and
knowledge in April 2007, and we intend to retain the services of
outside consultants, other than our independent registered
public accounting firm, with relevant accounting and audit
experience, skills and knowledge, working under the supervision
and direction of our management, to supplement our finance
personnel in the areas of external financial reporting,
corporate accounting and stock option accounting;
|
| |
| |
•
|
during 2007 we utilized outside consultants, other than our
independent registered public accounting firm, to assist our
management, working under its supervision and direction, in our
analysis and calculation of our income tax provision, and we
plan to continue to utilize outside consultants, other than our
independent registered public accounting firm, to assist our
management, working under its supervision and direction, in our
analysis of such matters in future periods;
|
| |
| |
•
|
with regard to any future material acquisition or partnership
that does not involve a well-known entity, management will
present a written report to our board of directors concerning
the proposed transaction, including a vetting of the management
team or practices of the third party;
|
| |
| |
•
|
we have improved month- and quarter-end closing procedures to
standardize our processes for financial review to ensure that
U.S. reviewers monitor financial information from
decentralized locations on a consistent manner and report their
findings to the Audit Committee and in 2008 we plan to continue
to enhance these procedures;
|
| |
| |
•
|
we have adopted a code of ethics for all directors, employees
and advisors in compliance with Nasdaq regulations;
|
| |
| |
•
|
we have adopted a whistleblower policy and are implementing
procedures that will allow for anonymous reporting of any
potential violations of law;
|
| |
| |
•
|
we have hired an experienced Chief Operating Officer to oversee
our day-to-day operations, further strengthening our commitment
to ensure accurate financial reporting, as well as compliance
with laws and regulations;
|
| |
| |
•
|
we have completed the review and reporting of two fiscal
quarters and the audits of fiscal years 2007, 2006, 2005 and
2004 in a timely manner and have filed one quarterly report on
Form 10-Q
and this annual report on
Form 10-K; and
|
| |
| |
•
|
we have formed a Disclosure Committee consisting of key
management members from operations, legal and finance, which
reviews all events during the quarter with the purpose that we
disclose all pertinent events in a timely and accurate manner.
|
Under the oversight of our audit committee, we are continuing to
review our processes and procedures to strengthen and improve
our internal controls, with the goals of ensuring accurate
financial reporting and complying with laws and regulations
applicable to us.
|
|
|
ITEM 9A.
|
Controls
and Procedures
|
(a) Evaluation of disclosure controls and
procedures. We maintain “disclosure controls
and procedures,” as such term is defined in
Rule 13a-15(e)
under the Securities Exchange Act of 1934 (the “Exchange
Act”), that are designed to ensure that information
required to be disclosed by us in reports that we file or submit
under the Exchange Act is recorded, processed, summarized, and
reported within the time periods specified in Securities and
Exchange Commission rules and forms, and that such information
is accumulated and communicated to our management, including our
chief executive officer and chief financial officer, as
appropriate, to allow timely decisions regarding required
disclosure. In designing and evaluating our disclosure controls
and procedures, management recognized that disclosure controls
and procedures, no matter how well conceived and operated, can
provide only reasonable, not absolute, assurance that the
objectives of the disclosure controls and procedures are met. In
response to comments from our auditors and our own
investigations, our disclosure controls and procedures have been
designed to meet, and management believes that they meet,
reasonable assurance standards. Additionally, in designing
disclosure controls and procedures, our management necessarily
was required to apply its
104
judgment in evaluating the cost-benefit relationship of possible
disclosure controls and procedures. The design of any disclosure
controls and procedures also is based in part upon certain
assumptions about the likelihood of future events, and there can
be no assurance that any design will succeed in achieving its
stated goals under all potential future conditions.
Based on their evaluation as of the end of the period covered by
this Annual Report on
Form 10-K,
our chief executive officer and chief financial officer have
concluded that, subject to the limitations noted above, our
disclosure controls and procedures were effective to ensure that
material information relating to us, including our consolidated
subsidiaries, is made known to them by others within those
entities, particularly during the period in which this Annual
Report on
Form 10-K
was being prepared.
(b) Changes in internal controls. During
the year ended March 31, 2007, we continued to make
improvements to our internal control structure and financial
reporting processes, including revising our authorization matrix
and our inventory control segregation policy in The Netherlands;
establishing fixed closing and reporting deadlines and fixed
budgeting and forecasting schedules; refining our procedures for
calculating and recording bad debt reserves and potential
revenue adjustments; establishing procedures for sell-through
method in The Netherlands; revising certain aspects of our
purchasing policy and procedures; conducting an actuarial study
of our social retirement funding in Mexico; and formalizing
procedures to ensure that all significant transactions undergo a
legal and accounting review and are reviewed and approved by our
board of directors. We have completed a risk assessment of the
significant financial statement accounts and developed a project
time lien and milestones for becoming Sarbanes-Oxley compliant
by the end of fiscal year 2008.
Other than these changes, there were no significant changes in
our internal control over financial reporting (as defined in
Rule 13a-15(f)
under the Exchange Act) identified in connection with the
evaluation described in Item 4(a) above that occurred
during our last fiscal year that has materially affected, or is
reasonably likely to materially affect, our internal control
over financial reporting.
This annual report does not include a report of
management’s assessment regarding internal control over
financial reporting or an attestation report of the
Company’s public accounting firm due to a transition period
established by rules of the Securities and Exchange Commission
for newly public companies.
|
|
|
ITEM 9B.
|
Other
Information
|
(a) 2008 Bonus Plan. On June 14,
2007, the compensation committee of the board of directors
approved a bonus plan for fiscal year 2008 (the “Bonus
Plan”), under which all of our employees, including our
executive officers, are eligible for bonus awards if we attain
specified targeted company goals and the individual obtains his
or her individual performance goals. Bonuses, if awarded, are
payable in cash or, at the determination of the Compensation
Committee no later than April 7, 2008 as necessary to
preserve our cash reserves, in part or in whole in grants of
stock options under our 2006 Stock Incentive Plan. Any stock
options will be valued by reference to the Black Scholes option
pricing calculation at the fair market value of our common stock
on June 7, 2008. However, the compensation committee may
not grant options exceeding the number of shares authorized
under the 2006 Stock Incentive Plan less the number of shares we
are contractually obligated to grant in the next year.
If the compensation committee determines to award one or more
bonuses, the amount of any employee’s bonus award under the
Bonus Plan will be conditioned on the company achieving
specified targeted revenue, expense, cash position and
operational goals, and on the employee achieving his or her
individual goals. The determination of whether the financial and
performance goals have been met will be in the sole discretion
of the compensation committee. In addition, the compensation
committee will establish and take into consideration additional
yearly operational goals, including milestones for clinical
trials, research and development, quality assurance and
corporate development, in determining whether any adjustment to
the target bonus should be adjusted upwards. Stretch bonuses
constitute an upper limit on the bonus potential for employees.
Under the Bonus Plan, no bonus is paid unless we meet specified
minimum revenue, operating expense and cash position targets for
fiscal 2008, and no bonus will be paid unless an
individual’s performance goals are achieved. The award of
any bonus, other than the guaranteed cash bonuses in the amount
of $50,000 each to two executive officers, and the bonuses for
fiscal year 2007 specified in the Bonus Plan, is at the sole
discretion of the compensation committee.
105
Upon achievement of minimum revenue, expense and cash position
targets and individual goals (the “Minimum Target”),
our employees (other than executive officers) are eligible to
earn a threshold award of 10% of their individual base salary or
additional stock option grants, at the discretion of the
compensation committee. Upon achievement of 100% of our revenue,
expense and cash position targets and certain of our operational
targets, and achievement of individual goals (the “Stretch
Target”), our employees (other than executive officers) are
eligible to earn a bonus award of up to 35% of their individual
base salary. If the compensation committee determines to award
one or more bonuses, and if Target Milestones are exceeded, but
Stretch Milestones are not achieved, the compensation committee
may, at its discretion, award a bonus in an amount between the
Target Milestone bonus and Stretch Milestone bonus amount.
Upon achievement of Minimum Target goals and Stretch Target
goals, the chief executive officer will be eligible to earn a
bonus of 100% to 200% of his individual base salary; and the
chief operating officer, the chief financial officer and the
vice president corporate development and general counsel will be
eligible to earn a bonus of 50% to 150% of his respective
individual base salary. The vice president operations and
international sales is eligible to receive up to $100,000 in
bonus for fiscal year 2008, which will be paid quarterly, if
awarded, and up to an additional $50,000 cash bonus payable at
the end of the fiscal year, based on achievement of certain
revenue and net operating income goals. We hired a vice
president regulatory and clinical trials who commenced his
employment on June 18, 2007, whose annual base salary is
$242,000. The compensation committee has authorized a guaranteed
cash bonus of $50,000 for fiscal year 2008 after the end of 2008.
The Bonus Plan also authorized the payment of a cash bonus in
the amount of $150,000 to our chief executive officer, cash
bonus in the amount of $60,000 to our vice president corporate
development and general counsel, and the immediate grant of an
option to purchase 150,000 shares of our common stock to
our chief operating officer, an option to purchase 25,000 of our
common stock to our vice president operations and international
sales, and an option to purchase 100,000 shares of our
common stock to our vice president corporate development and
general counsel, in each case based upon each employee’s
and the Company’s performance in fiscal year 2007. Each
option will be granted at the fair market value of our common
stock on the date of grant.
The summary of the Bonus Plan set forth above does not purport
to be complete and is qualified in its entirety by reference to
the Bonus Plan.
(b) Stock Dividend. On February 20,
20007 our board of directors authorized payment of a stock
dividend to the persons who were holders of the Series A
convertible preferred stock immediately prior to the close of
the or initial public offering. We issued 87,494 shares of
common stock in payment of the dividend on March 21, 2007.
In connection with the dividend, the Company’s net loss
available to common stockholders increased $404,000 and $121,000
in the years ended March 31, 2007, and 2006, respectively.
PART III
|
|
|
ITEM 10.
|
Directors
and Executive Officers of Registrant
|
The information required by this Item is incorporated by
reference to the definitive proxy statement for our 2007 Annual
Meeting of Stockholders to be filed with the Securities and
Exchange Commission within 120 days after the end of our
March 31, 2007 fiscal year (the “2007 Proxy
Statement”).
Item 405 of
Regulation S-K
calls for disclosure of any known late filing or failure by an
insider to file a report required by Section 16(a) of the
Exchange Act. This disclosure is contained in the section
entitled “Section 16(a) Beneficial Ownership Reporting
Compliance” in the Proxy Statement and is incorporated
herein by reference.
We have adopted a Code of Business Conduct that applies to all
of our officers and employees, including our chief executive
officer, president and chief operating officer, chief financial
officer and other employees who perform financial or accounting
functions. The Code of Business Conduct sets forth the basic
principles that guide the business conduct of our employees. We
have also adopted a Senior Financial Officers’ Code of
Ethics that specifically applies to our chief executive officer,
president and chief operating officer, chief financial officer,
and key management employees. Stockholders may request a free
copy of our Code of Business Conduct and Ethics and
106
our Senior Financial Officers’ Code of Ethics by contacting
Oculus Innovative Sciences, Inc., Attention: CFO,
1129 N. McDowell Blvd., Petaluma, California 94954.
To date, there have been no waivers under our Code of Business
Conduct and Ethics or Senior Financial Officers’ Code of
Ethics. We intend to disclose future amendments to certain
provisions of our Code of Business Conduct and Ethics or Senior
Officers’ Code of Ethics or any waivers, if and when
granted, of our Code of Business Conduct and Ethics or Senior
Officers’ Code of Ethics on our website at
http://www.oculusis.com
within four business days following the date of such
amendment or waiver.
Our board of directors has appointed an audit committee,
comprised of Mr. Richard Conley, as chairman, Mr. Jay
Birnbaum and Mr. James Schutz. The Board of Directors has
determined that Mr. Conley qualifies as an audit committee
financial expert under the definition outlined by the Securities
and Exchange Commission. In addition, Mr. Birnbaum
qualifies as an “independent director” under the
current rules of the NASDAQ Global Market and Securities and
Exchange Commission rules and regulations, and we intend to
replace Mr. Schutz with a person who qualifies as an
“independent director” prior to the expiration of the
phase-in period of the NASDAQ Marketplace Rules and the rules
under the Exchange Act.
|
|
|
ITEM 11.
|
Executive
Compensation
|
The information required by this Item is incorporated by
reference to the 2007 Proxy Statement.
|
|
|
ITEM 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters
|
The information required by this Item is incorporated by
reference to the 2007 Proxy Statement.
Information about securities authorized for issuance under our
equity compensation plans appears under the caption “Equity
Compensation Plan Information” in the Proxy Statement. That
portion of the Proxy Statement is incorporated by reference into
this report.
|
|
|
ITEM 13.
|
Certain
Relationships and Related Transactions
|
The information required by this Item is incorporated by
reference to the 2007 Proxy Statement.
|
|
|
ITEM 14.
|
Principal
Accountant Fees and Services
|
The information required by this Item is incorporated by
reference to the 2007 Proxy Statement.
|
|
|
ITEM 15.
|
Exhibits,
Financial Statement Schedules
|
(a) Documents filed as part of this report
(1) Financial Statements
Reference is made to the Index to Consolidated Financial
Statements of Oculus Innovative Sciences, Inc. under Item 8
of Part II hereof.
(2) Financial Statement Schedules
Financial statement schedules have been omitted that are not
applicable or not required or because the information is
included elsewhere in the Consolidated Financial Statements or
the Notes thereto. One additional schedule pursuant to
Article 5 required by
Rule 5-04,
Valuation and Qualifying Accounts Schedule will be filed by
amendment within 30 days.
(3) Exhibits
See Item 15(b) below. Each management contract or
compensatory plan or arrangement required to be filed has been
identified.
(b) Exhibits
107
Exhibit Index
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
3
|
.1(i)*
|
|
Restated Certificate of
Incorporation of Registrant.
|
|
|
3
|
.1(ii)
|
|
Form of Bylaws of Registrant
(incorporated by reference to exhibit 3.8 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.1
|
|
Specimen Common Stock Certificate
(incorporated by reference to the exhibit of the same number
filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.2
|
|
Warrant to Purchase Series A
Preferred Stock of Registrant by and between Registrant and
Venture Lending & Leasing III, Inc., dated
April 21, 2004 (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.3
|
|
Warrant to Purchase Series B
Preferred Stock of Registrant by and between Registrant and
Venture Lending & Leasing IV, Inc., dated
June 14, 2006 (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.4
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.5
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.6
|
|
Amended and Restated Investors
Rights Agreement, effective as of September 14, 2006
(incorporated by reference to the exhibit of the same number
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.7
|
|
Form of Promissory Note issued to
Venture Lending & Leasing III, Inc. (incorporated by
reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.8
|
|
Form of Promissory Note (Equipment
and Soft Cost Loans) issued to Venture Lending &
Leasing IV, Inc. (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.9
|
|
Form of Promissory Note (Growth
Capital Loans) issued to Venture Lending & Leasing IV,
Inc. (incorporated by reference to the exhibit of the same
number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.10
|
|
Form of Promissory Note (Working
Capital Loans) issued to Venture Lending & Leasing IV,
Inc. (incorporated by reference to the exhibit of the same
number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.11
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.12
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.1
|
|
Form of Indemnification Agreement
between Registrant and its officers and directors (incorporated
by reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.2
|
|
1999 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
108
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.3
|
|
2000 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.4
|
|
2003 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.5
|
|
2004 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.6
|
|
Form of 2006 Stock Incentive Plan
and related form stock option plan agreements (incorporated by
reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.7*
|
|
2006 Stock Incentive Plan Notice
of Stock Unit Award and Stock and Stock Unit Agreement issued to
Robert Miller
|
|
|
10
|
.8
|
|
Office Lease Agreement, dated
October 26, 1999, between Registrant and RNM Lakeville,
L.P. (incorporated by reference to exhibit 10.7 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.9
|
|
Amendment to Office Lease
No. 1, dated September 15, 2000, between Registrant
and RNM Lakeville L.P. (incorporated by reference to
exhibit 10.8 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.10
|
|
Amendment to Office Lease
No. 2, dated July 29, 2005, between Registrant and RNM
Lakeville L.P. (incorporated by reference to exhibit 10.9
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.11
|
|
Amendment No. 3 to Lease,
dated August 23, 2006, between Registrant and RNM Lakeville
L.P. (incorporated by reference to exhibit 10.23 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.12
|
|
Office Lease Agreement, dated
May 15, 2005, between Oculus Technologies of Mexico, S.A.
de C.V. and Antonio Sergio Arturo Fernandez Valenzuela
(translated from Spanish) (incorporated by reference to
exhibit 10.10 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.13
|
|
Office Lease Agreement, dated July
2003, between Oculus Innovative Sciences, B.V. and Artikona
Holding B.V. (translated from Dutch) (incorporated by reference
to exhibit 10.11 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.14
|
|
Loan and Security Agreement, dated
March 25, 2004, between Registrant and Venture
Lending & Leasing III, Inc. (incorporated by reference
to exhibit 10.12 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.15
|
|
Loan and Security Agreement, dated
June 14, 2006, between Registrant and Venture
Lending & Leasing IV, Inc. (incorporated by reference
to exhibit 10.13 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.16*
|
|
Amendment No. 1 to Supplement
to Loan and Security Agreement, dated March 29, 2007,
between Registrant and Venture Lending & Leasing IV,
Inc.
|
|
|
10
|
.17
|
|
Employment Agreement, dated
January 1, 2004, between Registrant and Hojabr Alimi
(incorporated by reference to exhibit 10.14 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.18
|
|
Employment Agreement, dated
January 1, 2004, between Registrant and Jim Schutz
(incorporated by reference to exhibit 10.15 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.19
|
|
Employment Agreement, dated
June 1, 2004, between Registrant and Robert Miller
(incorporated by reference to exhibit 10.16 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
109
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.20
|
|
Employment Agreement, dated
June 1, 2005, between Registrant and Bruce Thornton
(incorporated by reference to exhibit 10.17 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.21
|
|
Employment Agreement, dated
June 10, 2006, between Registrant and Mike Wokasch
(incorporated by reference to exhibit 10.19 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.22
|
|
Form of Director Agreement
(incorporated by reference to exhibit 10.20 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.23
|
|
Consultant Agreement, dated
October 1, 2005, by and between Registrant and White Moon
Medical (incorporated by reference to exhibit 10.21 filed
with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.24
|
|
Leasing Agreement, dated
May 5, 2006, made by and between Mr. Jose Alfonzo I.
Orozco Perez and Oculus Technologies of Mexico, S.A. de C.V.
(incorporated by reference to exhibit 10.22 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.25
|
|
Stock Purchase Agreement, dated
June 16, 2005, between Registrant, Quimica Pasteur, S de
R.L., Francisco Javier Orozco Gutierrez and Jorge Paulino
Hermosillo Martin (incorporated by reference to
exhibit 10.24 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.26
|
|
Framework Agreement, dated
June 16, 2005, between Javier Orozco Gutierrez, Quimica
Pasteur, S de R.L., Jorge Paulino Hermosillo Martin, Registrant
and Oculus Technologies de Mexico, S.A. de C.V. (incorporated by
reference to exhibit 10.25 filed with Registration
Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.27
|
|
Mercantile Consignment Agreement,
dated June 16, 2005, between Oculus Technologies de Mexico,
S.A. de C.V., Quimica Pasteur, S de R.L. and Francisco Javier
Orozco Gutierrez (incorporated by reference to
exhibit 10.26 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.28
|
|
Partnership Interest Purchase
Option Agreement, dated June 16, 2005, between Registrant
and Javier Orozco Gutierrez (incorporated by reference to
exhibit 10.27 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.29
|
|
Termination of Registrant and
Oculus Technologies de Mexico, S.A. de C.V. Agreements with
Quimica Pasteur, S de R.L. by Jorge Paulino Hermosillo Martin
(translated from Spanish) (incorporated by reference to
exhibit 10.28 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.30
|
|
Termination of Registrant and
Oculus Technologies de Mexico, S.A. de C.V. Agreements with
Quimica Pasteur, S de R.L. by Francisco Javier Orozco Gutierrez
(translated from Spanish) (incorporated by reference to
exhibit 10.29 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.31
|
|
Loan and Security Agreement, dated
November 7, 2006, between Registrant and Robert Burlingame
(incorporated by reference to exhibit 10.30 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.32
|
|
Non-Negotiable Secured Promissory
Note, dated November 10, 2006, between Registrant and
Robert Burlingame (incorporated by reference to
exhibit 10.31 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.33*
|
|
Amendment No. 1 to
Non-Negotiable Secured Promissory Note, dated March 29,
2007, between Registrant and Robert Burlingame.
|
|
|
10
|
.34
|
|
Subordination Agreement, dated
November 7, 2006, by and among Registrant, Robert
Burlingame, Venture Lending & Leasing III, LLC, and
Venture Lending & Leasing IV, LLC (incorporated by
reference to exhibit 10.32 filed with Registration
Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
110
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.35*
|
|
Amendment No. 1 to
Subordination Agreement, dated March 29, 2007, by and among
Registrant, Robert Burlingame, Venture Lending &
Leasing III, LLC, and Venture Lending & Leasing IV,
LLC.
|
|
|
10
|
.36
|
|
Consulting Agreement, effective
November 9, 2006, by and between Registrant and Robert
Burlingame (incorporated by reference to exhibit 10.33
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.37
|
|
Director Agreement, dated
November 8, 2006, by and between Registrant and Robert
Burlingame (incorporated by reference to exhibit 10.34
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.38†
|
|
Exclusive Marketing Agreement,
dated December 5, 2005, by and between Registrant and Alkem
Laboratories Ltd (incorporated by reference to
exhibit 10.35 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.39
|
|
Settlement Agreement, effective
September 21, 2006, by and among Registrant and
Messrs. Jorge Ahumada Ayala and Fernando Ahumada Ayala
(incorporated by reference to exhibit 10.36 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.40
|
|
Settlement Agreement, dated
October 25, 2006, by and between Registrant and
Mr. Kim Kelderman (incorporated by reference to
exhibit 10.37 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
21
|
.1*
|
|
List of Subsidiaries.
|
|
|
23
|
.1*
|
|
Consent of Marcum &
Kliegman LLP, independent registered public accounting firm.
|
|
|
24
|
.1*
|
|
Power of Attorney (see
pages 112-113
of this
Form 10-K).
|
|
|
31
|
.1*
|
|
Certification of Chief Executive
Officer pursuant to Securities Exchange Act
Rule 13a — 14(a), as adopted pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
31
|
.2*
|
|
Certification of Chief Financial
Officer pursuant to Securities Exchange Act
Rule 13a — 14(a), as adopted pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.1**
|
|
Certification of Chief Executive
Officer pursuant to 18 U.S.C. Section 1250, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.2**
|
|
Certification of Chief Financial
Officer pursuant to 18 U.S.C. Section 1250, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
* |
|
Filed herewith. |
| |
|
** |
|
The material contained in Exhibits 32.1 and 32.2 shall not
be deemed “filed” with the SEC and is not to be
incorporated by reference into any filing of the Company under
the Securities Act of 1933 or the Securities Exchange Act of
1934, whether made before or after the date hereof irrespective
of any general incorporation language contained in such filing,
except to the extent that Registrant specifically incorporates
it by reference. |
| |
|
† |
|
Confidential treatment has been granted with respect to certain
portions of this agreement. |
Copies of above exhibits not contained herein are available to
any stockholder, upon payment of a reasonable per page fee, upon
written request to: Chief Financial Officer, Oculus Innovative
Sciences, Inc., 1129 N. McDowell Blvd., Petaluma,
California 94954.
(c) Financial Statements and Schedules
Reference is made to Item 15(a)(2) above.
111
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this report to be signed on its behalf by the undersigned,
thereunto duly authorized.
OCULUS INNOVATIVE SCIENCES, INC.
Hojabr Alimi
President, Chief Executive Officer and
Chairman of the Board
(Principal Executive Officer)
Date: June 20, 2007
POWER OF
ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below constitutes and appoints Hojabr Alimi
and James J. Schutz, and each of them, his true and lawful
attorneys-in-fact, each with full power of substitution, for him
or her in any and all capacities, to sign any amendments to this
report on
Form 10-K
and to file the same, with exhibits thereto and other documents
in connection therewith, with the Securities and Exchange
Commission, hereby ratifying and confirming all that each of
said attorneys-in-fact or their substitute or substitutes may do
or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the registrant and in the capacities and on the
dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
/s/ Hojabr
Alimi
Hojabr
Alimi
|
|
President, Chief Executive Officer
and
Chairman of the Board
(Principal Executive Officer)
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Robert
E. Miller
Robert
E. Miller
|
|
Chief Financial Officer (Principal
Financial Officer and Principal Accounting Officer)
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Akihisa
Akao
Akihisa
Akao
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Jay
Edward Birnbaum
Jay
Edward Birnbaum
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Edward
M. Brown
Edward
M. Brown
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Richard
Conley
Richard
Conley
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Gregory
M. French
Gregory
M. French
|
|
Director
|
|
June 20, 2007
|
112
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
/s/ James
J. Schutz
James
J. Schutz
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Robert
Burlingame
Robert
Burlingame
|
|
Director
|
|
June 20, 2007
|
|
|
|
|
|
|
|
/s/ Hojabr
Alimi
Hojabr
Alimi
|
|
Attorney-in-fact
|
|
June 20, 2007
|
113
EXHIBIT INDEX
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
3
|
.1(i)*
|
|
Restated Certificate of
Incorporation of Registrant.
|
|
|
3
|
.1(ii)
|
|
Form of Bylaws of Registrant
(incorporated by reference to exhibit 3.8 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.1
|
|
Specimen Common Stock Certificate
(incorporated by reference to the exhibit of the same number
filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.2
|
|
Warrant to Purchase Series A
Preferred Stock of Registrant by and between Registrant and
Venture Lending & Leasing III, Inc., dated
April 21, 2004 (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.3
|
|
Warrant to Purchase Series B
Preferred Stock of Registrant by and between Registrant and
Venture Lending & Leasing IV, Inc., dated
June 14, 2006 (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.4
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.5
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.6
|
|
Amended and Restated Investors
Rights Agreement, effective as of September 14, 2006
(incorporated by reference to the exhibit of the same number
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.7
|
|
Form of Promissory Note issued to
Venture Lending & Leasing III, Inc. (incorporated by
reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.8
|
|
Form of Promissory Note (Equipment
and Soft Cost Loans) issued to Venture Lending &
Leasing IV, Inc. (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.9
|
|
Form of Promissory Note (Growth
Capital Loans) issued to Venture Lending & Leasing IV,
Inc. (incorporated by reference to the exhibit of the same
number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.10
|
|
Form of Promissory Note (Working
Capital Loans) issued to Venture Lending & Leasing IV,
Inc. (incorporated by reference to the exhibit of the same
number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.11
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
4
|
.12
|
|
Form of Warrant to Purchase Common
Stock of Registrant (incorporated by reference to the exhibit of
the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.1
|
|
Form of Indemnification Agreement
between Registrant and its officers and directors (incorporated
by reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.2
|
|
1999 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.3
|
|
2000 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.4
|
|
2003 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.5
|
|
2004 Stock Plan and related form
stock option plan agreements (incorporated by reference to the
exhibit of the same number filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.6
|
|
Form of 2006 Stock Incentive Plan
and related form stock option plan agreements (incorporated by
reference to the exhibit of the same number filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.7*
|
|
2006 Stock Incentive Plan Notice
of Stock Unit Award and Stock and Stock Unit Agreement issued to
Robert Miller
|
|
|
10
|
.8
|
|
Office Lease Agreement, dated
October 26, 1999, between Registrant and RNM Lakeville,
L.P. (incorporated by reference to exhibit 10.7 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.9
|
|
Amendment to Office Lease
No. 1, dated September 15, 2000, between Registrant
and RNM Lakeville L.P. (incorporated by reference to
exhibit 10.8 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.10
|
|
Amendment to Office Lease
No. 2, dated July 29, 2005, between Registrant and RNM
Lakeville L.P. (incorporated by reference to exhibit 10.9
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.11
|
|
Amendment No. 3 to Lease,
dated August 23, 2006, between Registrant and RNM Lakeville
L.P. (incorporated by reference to exhibit 10.23 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.12
|
|
Office Lease Agreement, dated
May 15, 2005, between Oculus Technologies of Mexico, S.A.
de C.V. and Antonio Sergio Arturo Fernandez Valenzuela
(translated from Spanish) (incorporated by reference to
exhibit 10.10 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.13
|
|
Office Lease Agreement, dated July
2003, between Oculus Innovative Sciences, B.V. and Artikona
Holding B.V. (translated from Dutch) (incorporated by reference
to exhibit 10.11 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.14
|
|
Loan and Security Agreement, dated
March 25, 2004, between Registrant and Venture
Lending & Leasing III, Inc. (incorporated by reference
to exhibit 10.12 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.15
|
|
Loan and Security Agreement, dated
June 14, 2006, between Registrant and Venture
Lending & Leasing IV, Inc. (incorporated by reference
to exhibit 10.13 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.16*
|
|
Amendment No. 1 to Supplement
to Loan and Security Agreement, dated March 29, 2007,
between Registrant and Venture Lending & Leasing IV,
Inc.
|
|
|
10
|
.17
|
|
Employment Agreement, dated
January 1, 2004, between Registrant and Hojabr Alimi
(incorporated by reference to exhibit 10.14 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.18
|
|
Employment Agreement, dated
January 1, 2004, between Registrant and Jim Schutz
(incorporated by reference to exhibit 10.15 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.19
|
|
Employment Agreement, dated
June 1, 2004, between Registrant and Robert Miller
(incorporated by reference to exhibit 10.16 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.20
|
|
Employment Agreement, dated
June 1, 2005, between Registrant and Bruce Thornton
(incorporated by reference to exhibit 10.17 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.21
|
|
Employment Agreement, dated
June 10, 2006, between Registrant and Mike Wokasch
(incorporated by reference to exhibit 10.19 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.22
|
|
Form of Director Agreement
(incorporated by reference to exhibit 10.20 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.23
|
|
Consultant Agreement, dated
October 1, 2005, by and between Registrant and White Moon
Medical (incorporated by reference to exhibit 10.21 filed
with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.24
|
|
Leasing Agreement, dated
May 5, 2006, made by and between Mr. Jose Alfonzo I.
Orozco Perez and Oculus Technologies of Mexico, S.A. de C.V.
(incorporated by reference to exhibit 10.22 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.25
|
|
Stock Purchase Agreement, dated
June 16, 2005, between Registrant, Quimica Pasteur, S de
R.L., Francisco Javier Orozco Gutierrez and Jorge Paulino
Hermosillo Martin (incorporated by reference to
exhibit 10.24 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.26
|
|
Framework Agreement, dated
June 16, 2005, between Javier Orozco Gutierrez, Quimica
Pasteur, S de R.L., Jorge Paulino Hermosillo Martin, Registrant
and Oculus Technologies de Mexico, S.A. de C.V. (incorporated by
reference to exhibit 10.25 filed with Registration
Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.27
|
|
Mercantile Consignment Agreement,
dated June 16, 2005, between Oculus Technologies de Mexico,
S.A. de C.V., Quimica Pasteur, S de R.L. and Francisco Javier
Orozco Gutierrez (incorporated by reference to
exhibit 10.26 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.28
|
|
Partnership Interest Purchase
Option Agreement, dated June 16, 2005, between Registrant
and Javier Orozco Gutierrez (incorporated by reference to
exhibit 10.27 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.29
|
|
Termination of Registrant and
Oculus Technologies de Mexico, S.A. de C.V. Agreements with
Quimica Pasteur, S de R.L. by Jorge Paulino Hermosillo Martin
(translated from Spanish) (incorporated by reference to
exhibit 10.28 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.30
|
|
Termination of Registrant and
Oculus Technologies de Mexico, S.A. de C.V. Agreements with
Quimica Pasteur, S de R.L. by Francisco Javier Orozco Gutierrez
(translated from Spanish) (incorporated by reference to
exhibit 10.29 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.31
|
|
Loan and Security Agreement, dated
November 7, 2006, between Registrant and Robert Burlingame
(incorporated by reference to exhibit 10.30 filed with
Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.32
|
|
Non-Negotiable Secured Promissory
Note, dated November 10, 2006, between Registrant and
Robert Burlingame (incorporated by reference to
exhibit 10.31 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.33*
|
|
Amendment No. 1 to
Non-Negotiable Secured Promissory Note, dated March 29,
2007, between Registrant and Robert Burlingame.
|
|
|
10
|
.34
|
|
Subordination Agreement, dated
November 7, 2006, by and among Registrant, Robert
Burlingame, Venture Lending & Leasing III, LLC, and
Venture Lending & Leasing IV, LLC (incorporated by
reference to exhibit 10.32 filed with Registration
Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.35*
|
|
Amendment No. 1 to
Subordination Agreement, dated March 29, 2007, by and among
Registrant, Robert Burlingame, Venture Lending &
Leasing III, LLC, and Venture Lending & Leasing IV,
LLC.
|
|
|
10
|
.36
|
|
Consulting Agreement, effective
November 9, 2006, by and between Registrant and Robert
Burlingame (incorporated by reference to exhibit 10.33
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.37
|
|
Director Agreement, dated
November 8, 2006, by and between Registrant and Robert
Burlingame (incorporated by reference to exhibit 10.34
filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Description
|
|
|
|
|
10
|
.38†
|
|
Exclusive Marketing Agreement,
dated December 5, 2005, by and between Registrant and Alkem
Laboratories Ltd (incorporated by reference to
exhibit 10.35 filed with Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.39
|
|
Settlement Agreement, effective
September 21, 2006, by and among Registrant and
Messrs. Jorge Ahumada Ayala and Fernando Ahumada Ayala
(incorporated by reference to exhibit 10.36 filed with
Registration Statement on
Form S-1
(File
No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
10
|
.40
|
|
Settlement Agreement, dated
October 25, 2006, by and between Registrant and
Mr. Kim Kelderman (incorporated by reference to
exhibit 10.37 filed with Registration Statement on
Form S-1
(File No. 333-135584),
as amended, declared effective on January 24, 2007).
|
|
|
21
|
.1*
|
|
List of Subsidiaries.
|
|
|
23
|
.1*
|
|
Consent of Marcum &
Kliegman LLP, independent registered public accounting firm.
|
|
|
24
|
.1*
|
|
Power of Attorney (see
pages 112-113
of this
Form 10-K).
|
|
|
31
|
.1*
|
|
Certification of Chief Executive
Officer pursuant to Securities Exchange Act
Rule 13a — 14(a), as adopted pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
31
|
.2*
|
|
Certification of Chief Financial
Officer pursuant to Securities Exchange Act
Rule 13a — 14(a), as adopted pursuant to
Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.1**
|
|
Certification of Chief Executive
Officer pursuant to 18 U.S.C. Section 1250, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32
|
.2**
|
|
Certification of Chief Financial
Officer pursuant to 18 U.S.C. Section 1250, as adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
* |
|
Filed herewith. |
| |
|
** |
|
The material contained in Exhibits 32.1 and 32.2 shall not
be deemed “filed” with the SEC and is not to be
incorporated by reference into any filing of the Company under
the Securities Act of 1933 or the Securities Exchange Act of
1934, whether made before or after the date hereof irrespective
of any general incorporation language contained in such filing,
except to the extent that Registrant specifically incorporates
it by reference. |
| |
|
† |
|
Confidential treatment has been granted with respect to certain
portions of this agreement. |